Entering edit mode

5.7 years ago
tanya_fiskur
▴
80
Hi everyone!
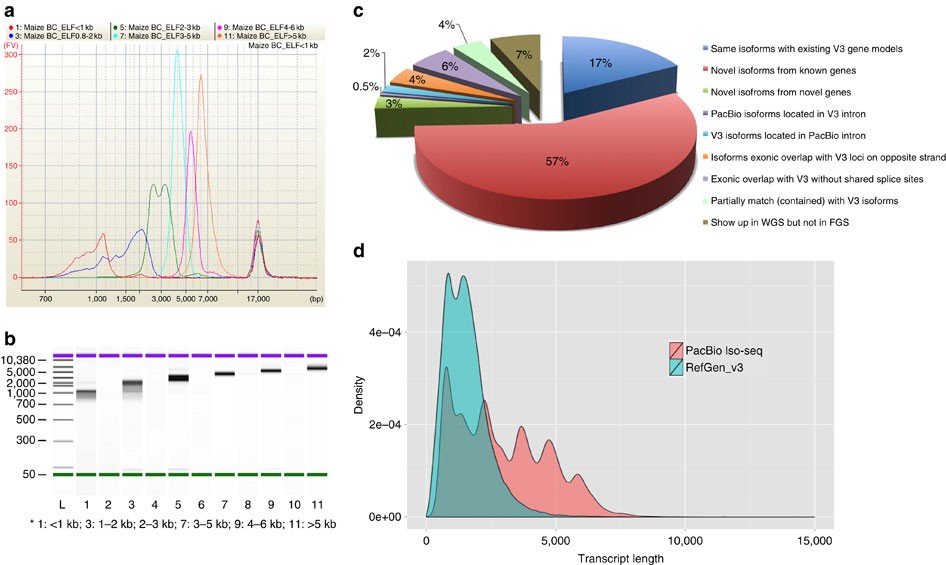
I am really wondering, with which tool is it possible to make a plot of transcript length distribution, based on the genome + gene annotation, like it was done here (figure D).
In the article itself and in the supplementary materials it is not mentioned.
Thanks very much in advance!


The paper clearly states that the PacBio Iso-seq data is available:
If you want to use the PacBio data you would need to download and process that raw data according to their methods (unless they have prebuilt uploaded annotations somewhere else - or maybe you could ask the PI directly?)
Thank you very much for the code! I have another pacbio data and gtf annotation, and want to create a similar plot. Probably, the gffcompare output can give me the lengths of the transcripts.
Another option if you are using R (similar to my code) is to use your GTF file to produce a TranscriptDb object similar to the package-derived one used in my code.
Thank you! And does the transcriptsBy function work with the gtf-derieved file? Also, did I understand you correctly that you split the data in two halves just to show two overlapping plots, these is no other reason to do it?