
Info: 28.6.22
This tutorial is all-deprecated. Use sra-explorer.info to get download links from ENA and Setting up Aspera Connect (ascp) on Linux and macOS to learn how to setup Aspera.
Info: 16.05.2021
The layout of ENA changed so the steps in this tutorial towards finding and selecting datasets are deprecated, and I have not yet found the time to update it. Actually, as there is sra-explorer these days I recommend to just use this to query ENA (or ncbi), see below.
Edit 06.05.20: I actually recommend to use sra-explorer these days to get download links. This is the most convenient and fastest way. It implements the creation of download links from both NCBI and ENA including links for Aspera-driven downloads based on this tutorial. Step 1 of this tutorial can help setting up Aspera. The other steps are redundant if using sra-explorer, but this tutorial has been written before that tool was published. For more details see also sra-explorer : find SRA and FastQ download URLs in a couple of clicks and visit sra-explorer.info. It is self-explanatory.
TUTORIAL
Published sequencing data are commonly stored at NCBI and questions on how to efficiently download these data are posted frequently at Biostars. While NCBI relies on its SRA format to share these data it is possible to directly download them in fastq format from the European Nucleotide Archive (ENA1) which mirrors NCBI.
In this tutorial, we will examplarily download an entire dataset of ChIP-seq and ATAC-seq data, requiring minimal preprocessing work. We will use the Aspera client for download rates of several tens of Mb/s up to few hundred Mb/s (depending on the connection, I/O capacity and distance to the download location). This example code should work on Linux and Mac.
Step-1: Get the Aspera client
Go to https://www.ibm.com/aspera/connect/ and get the most recent installer for your system.
For Linux, it is a tarball (use tar zxvf to unpack) with an installer batch script and for Mac, a standard disk image.
After installation, there now will be these executables/files in their default locations:
Linux:
$HOME/.aspera/connect/bin/ascp --- the executable
$HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh --- openssh file that we'll need later
Mac:
$HOME/Applications/Aspera\ Connect.app/Contents/Resources/ascp --- the executable
$HOME/Applications/Aspera\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh --- openssh file that we'll need later
In any case, make sure you add the folder with the ascpexecutable to your PATH.
If PATH is a new word to you, please google it ;-)
Step-2: Choose your dataset
We have multiple options here.
Use sra-explorer: The sra-explorer is a convenient GUI to get browse NCBI for datasets. In our case we enter our accession number PRJNA288801 into the search field, then select the desired samples, add them to the cart and then copy the Aspera download links it produces. I will not add further details here as the tool is self-explainatory. It includes many (download) options, check it out. Thanks to Phil Ewels for this awesome tool!
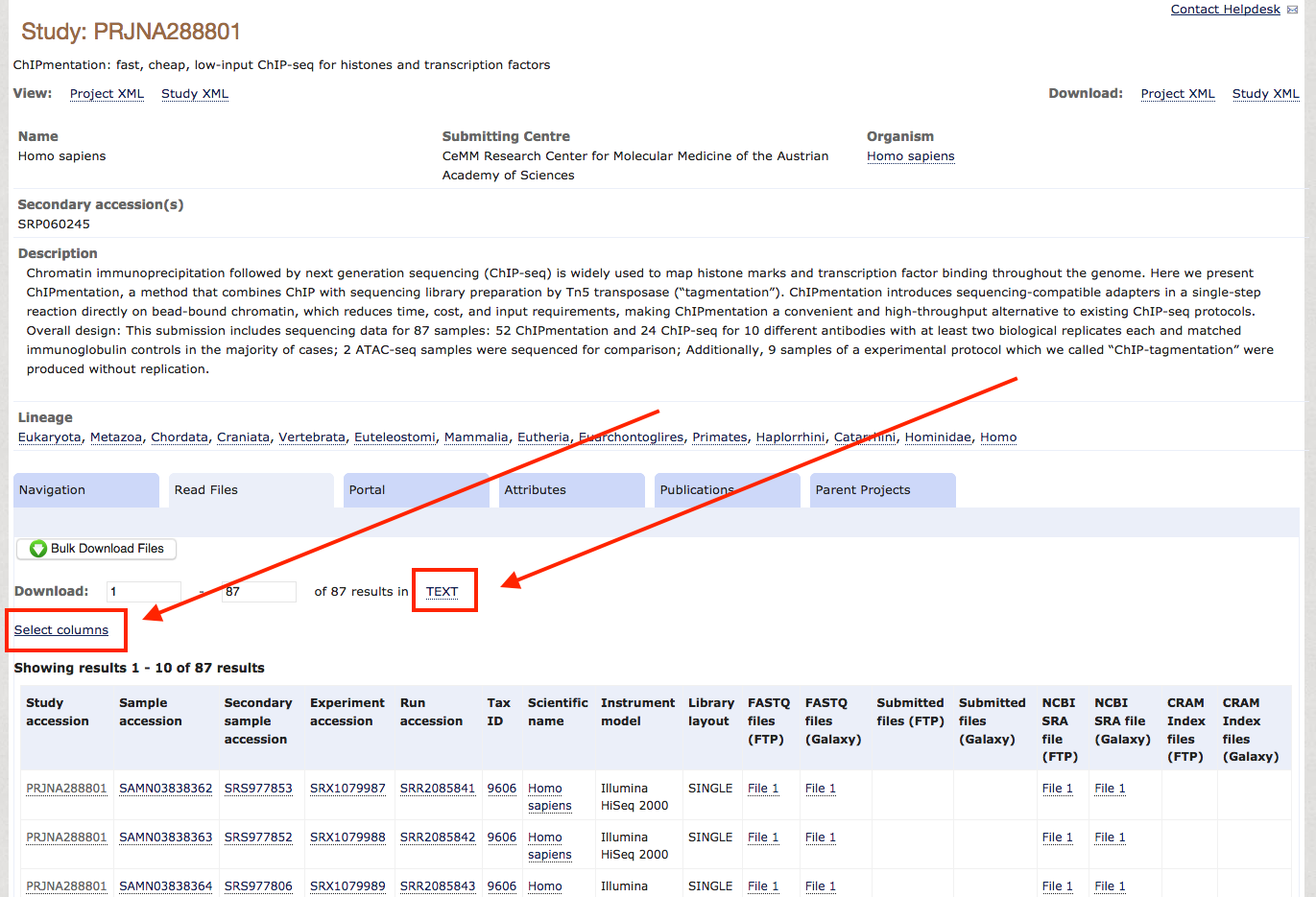
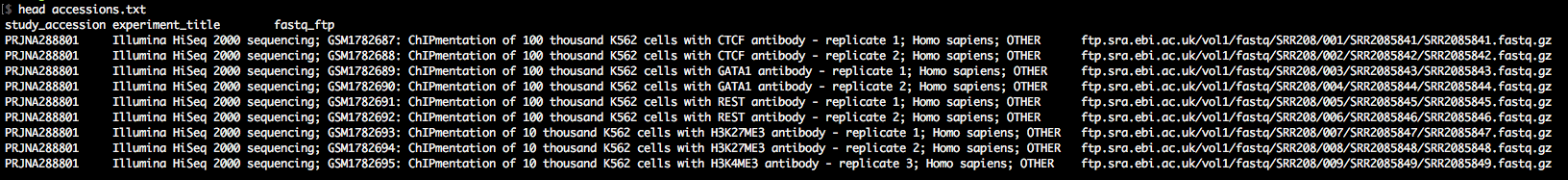
Alternatively, query ENA / NCBI manually to find datasets: Once you know which data you want to download, check if they are backed up on the ENA, which is true for most unrestricted data. For this tutorial, we will download the entire dataset from the ChIPmentation paper of 2015. When you check the paper for the NCBI accession, you'll find GSE70482. Following this link, you find the BioSample accession number PRJNA288801. So you go to the ENA, enter this PRJNA288801 in the search field and find a summary page with all available data for download. Scrolling down a bit, you see a table with accession numbers and all kinds of metadata. As typically we do not need most of these metadata, we use the field Select columns to select the essential metadata we need for the download, which are Study Accession, FASTQ files (FTP) and Experiment title. After selecting these, and unselecting everything else, you press TEXT and save the file as accessions.txt in your project folder.
Step-3: Download the data
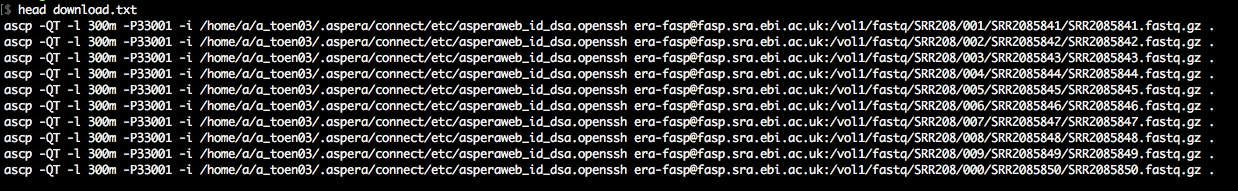
As you'll see in accessions.txt, the download paths direct you to the ENA ftp-server, which is rather slow. We want to download with the Aspera client (up to 200Mb/s at my workplace). Therefore, we awk around a bit to change the download paths to the era-fasp server. As you'll see in case of paired-end data, the paths to the two mate fastq files in accessions.txt
are separated by semicolon, which we take into account. The output of this snippet is download.txt.
Linux:
awk 'FS="\t", OFS="\t" { gsub("ftp.sra.ebi.ac.uk", "era-fasp@fasp.sra.ebi.ac.uk:"); print }' accessions.txt | cut -f3 | awk -F ";" 'OFS="\n" {print $1, $2}' | awk NF | awk 'NR > 1, OFS="\n" {print "ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh" " " $1 " ."}' > download.txt
Mac:
awk 'FS="\t", OFS="\t" { gsub("ftp.sra.ebi.ac.uk", "era-fasp@fasp.sra.ebi.ac.uk:"); print }' accessions.txt | cut -f3 | awk -F ";" 'OFS="\n" {print $1, $2}' | awk NF | awk 'NR > 1, OFS="\n" {print "ascp -QT -l 300m -P33001 -i $HOME/Applications/Aspera\\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh" " " $1 " ."}' > download.txt
The output is a simple list of download commands using ascp.
That's it. Now, we only have to run the download commands.
Edit (23.07.18): The download paths are always like era-fasp@fasp.sra.ebi.ac.uk:/vol1(...). I point that out because of a recent post (328182) where OP accidentally forgot the ":" after the .ac.uk and used fasp@ instead of era-fasp@.
Lets download:
## Either by a simple loop:
while read LIST; do
$LIST; done < download.txt
## or by using GNU parallel to have things parallelized:
cat download.txt | parallel "{}"
Once the download is complete, one can play around using the accessions.txt to rename the files with e.g. information from the Experiment title field (column 2), or other metadata you may retrieve from ENA.
Edit 28.2.19: For matters of completeness, I also add a suggestion on how to get the same data from NCBI using prefetch and parallel-fastq-dump, a wrapper for fastq-dump from Renan Valieris for parallelized fastq conversion from sra files. Say one has a file IDs.txt which contains the SRA file IDs like:
SRRXXXXXX1
SRRXXXXXX2
(...)
SRRXXXXXXn
one can use this simple function to download SRA files via prefetch, followed by fastq conversion with parallel-fastq-dump.
Note In the past prefetch could be coupled with Aspera for fast downloads, this is no longer the case, see https://github.com/ncbi/sra-tools/issues/255. Now prefetch will always download via the standard https connection.
function LoadDump {
prefetch -O ./ -X 999999999 $1
if [[ -e ${1}.sra ]]; then
parallel-fastq-dump -s ${1}.sra -t 8 -O ./ --tmpdir ./ --split-3 --gzip && rm ${1}.sra
else
echo '[ERROR]' $1 'apparently not successfully loaded' && exit 1
fi
}; export -f LoadDump
cat IDs.txt | parallel -j 2 "LoadDump {}"
This would use 8 threads for fastq conversion and run two SRA files at a time via GNU parallel, hence requiring 16 threads. As always, scale up or down based on the available resources and potential I/O bottlenecks on your system.



Good work! I once had the pleasure to use
fastq-dumpon whole-genome data. I was cursing in multiple languages! :DI recently was downloading the data form CCLE experiment and it was taking ages (also crushing more than once) with
sra-toolkitandfastq-dump. I used your approach, slightly it modifying and it worked wonders! Thanks a lot!My modification below:
where
${tissue_of_interest}and${experiment}where variables I set up specifically to my needs (i.e. CERVIX, RNA-seq). I copied this intosra-explorer. The Project has too many files to directly searchsra-explorerwith its ID.Glad to hear it is used productively :)
I am sorry. I am considering switch from SRA because the files I want to download are too big and I got unkown error using SRA for some time. I am trying to use aspera client to download the fastq files. From the link above I downloaded the ibm-aspera-connect-3.9.9.177872-linux-g2.12-64.tar.gz and decompressed it to ibm-aspera-connect-3.9.9.177872-linux-g2.12-64.sh.
I also set the path to the directory I am working:
everything looks fine until the next step;
Which part did I made the mistake
PATH is the path to the folder containing the binaries, not the binary itself, please try:
PATH=$PATH:/home/caiqi/.aspera/connect/bin/Thank you for explanation. It is working and it is faster than I expected
Good work dude. Will use this next time I need to get data from ENA.
Thank you Sir, glad to help!
I finally got this to work. Do you know why I could be getting variable download speeds? The frist time I ran this, I was getting speeds of ~230 MB/s and now I'm getting 1.7 MB/s at best. Is there an option I can choose to increase the speed? What could be affecting this you think?
Which of the method is usually the fastest to download fastq files? I know it depends a lot on network yet over a stable connection (but low bandwidth) which one of wget, curl, aspera or SRA toolkit performs better?
Aspera will be the fastest , instead of curl or wget use Aria2 for a faster download.
SRAtoolkit downloads via http but it performs some internal validation checks and only will produce the final SRA file on disk if download finished successfully afaik, so for downloads from SRA I prefer it. NCBI does not support Aspera anymore.
For ENA it would be Aspera, this is why I cover it as the default method in this tutorial. Still, unless you download hundreds of GB or even TB standard download via wget is fine as well. With our HPC I tyically get download rates via wget from ENA in the ~50-60Mb/s range. Sufficient for most applications unless you are going to download entire cohorts.