Entering edit mode

5.3 years ago
shadowstep
•
0
I followed the nCov course in biostar handbook, I did the following in command line using the nCov genomes from NCBI database:
nCov=NC_045512.fa
minimap2 -a $nCov nCov-genomes.fa | samtools sort > nCov-align.bam
# Index the alignments
samtools index nCov-align.bam
# Compute the genotypes from the alignment file with pileup.
bcftools mpileup -B -Ovu -f $nCov nCov-align.bam > nCov-genotypes.vcf
cat nCov-variants.vcf | bcftools query -f '%CHROM %POS %REF %ALT\n' > nCov-variants.txt
The results showed that there are variants at five positions
NC_045512 8782 C T
NC_045512 17747 C T
NC_045512 17858 A G
NC_045512 18060 C T
NC_045512 28144 T C
I also built and plotted a phylo tree for all these genomes using ggtree. Now If I want to use gheatmap() function to show the variants at each position and map it to the phylotree, how can I generate a genotype matrix that meets the requirement of gheatmap()?
I want to make a plot like this:
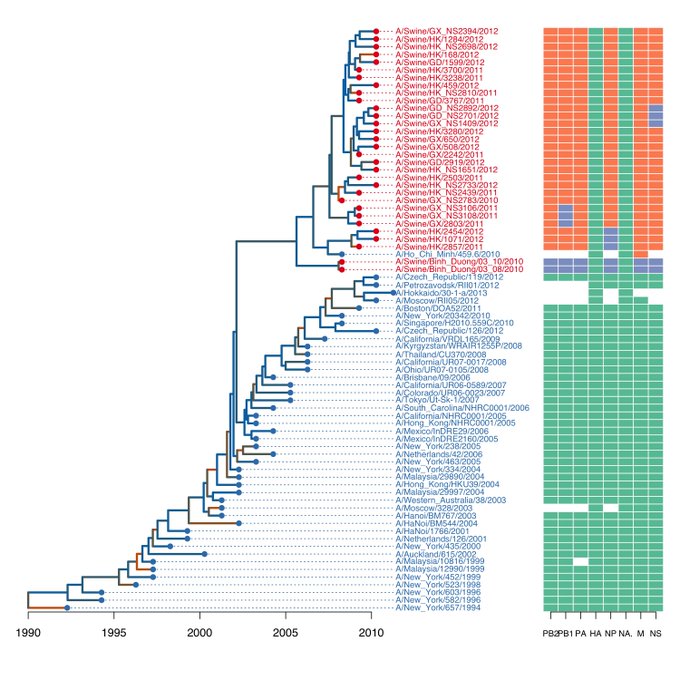
This function requires a genotype matrix like this:
PB2 PB1 PA HA NP NA M NS
A/Swine/Binh_Duong/03_10/2010 trig trig trig HuH3N2 trig HuH3N2 trig trig
A/Swine/Binh_Duong/03_08/2010 trig trig trig HuH3N2 trig HuH3N2 trig trig
A/Swine/HK/2857/2011 pdm pdm pdm HuH3N2 trig HuH3N2 pdm pdm
A/Swine/HK/1071/2012 pdm pdm pdm HuH3N2 trig HuH3N2 pdm pdm
A/Swine/HK/2454/2012 pdm pdm pdm HuH3N2 trig HuH3N2 pdm pdm
A/Swine/GX_NS2783/2010 pdm pdm pdm HuH3N2 pdm HuH3N2 pdm pdm
A/Swine/HK_NS2733/2012 pdm pdm pdm HuH3N2 pdm HuH3N2 pdm pdm
A/Swine/HK_NS2439/2011 pdm pdm pdm HuH3N2 pdm HuH3N2 pdm pdm
A/Swine/HK/2503/2011 pdm pdm pdm HuH3N2 pdm HuH3N2 pdm pdm
A/Swine/GX/2803/2011 pdm trig pdm HuH3N2 pdm HuH3N2 pdm pdm
A/Swine/GX_NS3108/2011 pdm trig pdm HuH3N2 pdm HuH3N2 pdm pdm
A/Swine/GX_NS3106/2011 pdm trig pdm HuH3N2 pdm HuH3N2 pdm pdm