Entering edit mode

4.5 years ago
marongiu.luigi
▴
730
Hello,
I performed a WGS with Illumina on three histological sections. I looked for viral genomes and I got one hit for Epstein-Barr virus. Then I performed PCR to confirm the data and I got an unclear result for the sample positive by WGS and two positives for the other two samples.
How shall I consider this analysis? That the WGS was garbage? Is there a way that PCR and WGS could give different values? for instance, the sections were different...
Thank you

I am not concerned about the one negative PCR but the two positive ones: they disprove the WGS analysis... I used a concatenated genome with all the viral sequences in GeneBank, added it to the human genome and aligned with BWA. But simple there are no reads.
Wouldn't simply putting together a lot of virus sequences create a lot of sequence-identical (or at least highly similar) contigs resulting in multimappers which in turn lead to no counts for all these sequence-identical regions? I cannot comment further since, as Joe says, your question lacks any details what you exactly did and what the samples were.
You probably need to tell us more about what 'positive' looks like in your WGS? How many reads? What coverage of the genome?
My bet is on multi mapping or something if the WGS is 'wrong', but more likely false positives in your PCR through contamination, mixed up samples, or something.
On IGV the positive (bottom alignment) looks like this: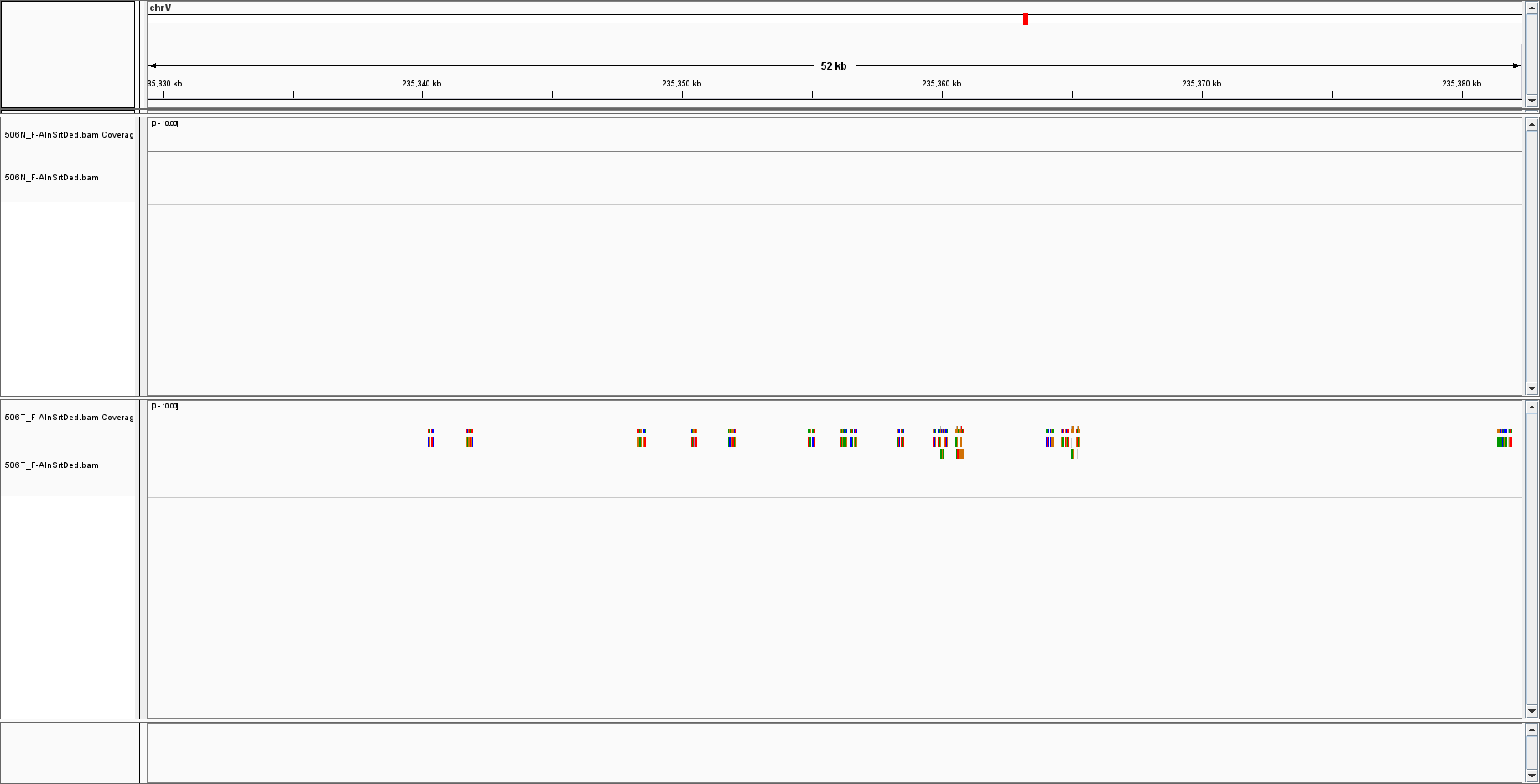 This sample had 10 reads about 100 bp each.
By extracting the EBV chromosome, I got these for the positive WGS / negative PCR sample:
This sample had 10 reads about 100 bp each.
By extracting the EBV chromosome, I got these for the positive WGS / negative PCR sample:
compared to a negative WGS / positive PCR:
It seems very unlikely that 10 reads reflects any sort of biological reality. These are probably multimappers/low quality reads or just happen to have some bits of sequence that are shared between viral motifs/human motifs.
I would suggest looking at the mapping file for those reads and considering whether they're garbage or not.