I am trying to analyze some ATAC seq data. I performed paired-end sequencing on six samples. Three samples are from condition A and three samples from condition B. I have called peaks using MACS2 and finding it confusing on how to submit these to Dseq2. Do I need to merge/intersect files before submitting to Dseq2? If so, how do I do that? Thanks
There is no gold standard for this. I personally use the Genrich peak caller to call peaks across the biological replicates for each condition. The tool accepts replicates and then produces a single peak list that represents the reproducible peaks per group. These I merge the peaks of all groups using bedtools merge, write to SAF format like awk 'OFS="\t" {print $1_"$2"_"$3, $1, $2, $3, "."}' merged.bed > merged.saf and then use featureCounts to produce a count matrix. The output will be a samples as columns and regions as rows matrix with the raw counts for every region per sample. This you can then read into R and put into any differential analysis tool such as DESeq2. Hope that helps.
Edit1: Genrich has the advantage that it takes replicates and then combines p-values for each of these into a single p-value using Fisher's method. That omits the need for post-hoc filtering and reduces spurious peak calls that are not reproducible between replicates. The main advantage over this in comparison to other filtering approaches is that 1) it does not require aritrary thresholds like "peak must be in 2/3 samples" and 2) it does not require the IDR framework which is commonly used as the current implementation only accepts n=2 even though every proper experiment should at least have a triplicate. Genrich is not published and therefore not peer reviewed so try it out, examine results on a genome browser and decide for yourself.
Edit2: The paper that was linked below mentions the PePr peak caller. This one also accepts replicates but requires an input control which is typically not available for ATAC-seq so don't even try this one.
Thanks ATpoint. Yes no 'input' was submitted for ATACseq so will be sticking with Genrich. Some follow up questions:
I used MACS2 previously which gave me six independent narrowpeak files, which I then plugged into ChIPseeker for peak annotation and visualization. However, if I now use Genrich, the output from my three biological replicates will be one narrowpeak file, how can I differentiate the three samples in the ChIPseeker package?
My three samples are from three individual patients and I would still like to know peak location independently.
Do the bam files need to be labeled in a certain way for Genrich, ie control1.bam, control2.bam, control3.bam and treat1.bam, treat2.bam, treat3.bam?
I am not a ChIPseeker user so I cannot comment.
You can run Genrich on each sample separately to get individual peaks. I personally find it more meaningful though only to use the consensus list per group for annotations. Individual peaks per patient might be interesting but you cannot know if these are indeed reproducible or sometimes simply a product of different data quality in different samples. This is the whole point of using a list of reproducible rather than individual peaks. Be sure you run Genrich on the controls and treats separately, not in the same analysis.
Hi ATpoint, can you please comment on my second question? To combine biological replicates, do I need to label bam files for Genrich in a certain way? Thanks
No, please read the manual of Genrich. You simply provide the BAM files you want to be called as one group. You would perform one run of peak calling with all samples of group1 and one with group2. The tool does not know what is inside the bam files, it just sees different files, will call peaks for each of it and then output a combined pvalue for every peak.
I have managed to call peaks using Genrich and subsequently merged peaks using bedtools merge. I then converted this file to SAF format for featureCounts. However, I am a little confused as to which command to use for featureCounts. I have seen you numerous suggestions in biostars posts and this seems to be the consensus:
featureCounts -p -a peaks.saf -F SAF -o out.txt atacseq.bam
I don't seem to understand what the bam file is. Also is the peaks.saf file in the above example an annotation file (seems like if you look at the featureCounts documentation) or is it the saf file generated from the merged peaks file.
Thanks.
The SAF is created from the merged peaks. BAM files are all bam files from every sample, so one bam per sample. Out all BAMs from your experiment into one folder and simply use *.bam
The same BAM files you used for peak calling, no specific sort order. Name-sorted is fine. I personally do not use -p since in my understanding each read (or rather read start site) in paired-end sequencing represents a unique cutting event from ATAC-seq. Using -p would throw away half of your observations imho.
Hi ATpoint,
I know this was written 9 weeks ago, but I was wondering if you could explain further why you do not use -p if the ATAC sequencing run was paired ended?
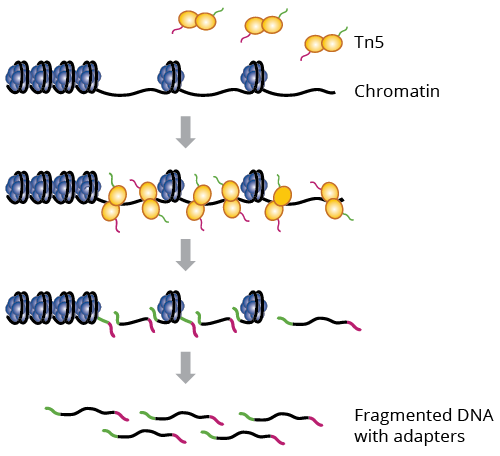
Based on my understanding the transposon (Tn) integrates into the genome and makes a single cut, introducing the sequencing adapter. This is what we detect as one end of a sequencing read. The second end of the read is the position where a second transposon integrated and introduced its adapter. Therefore I prefer to count cutting events (which is the end of each read) rather than fragments because the two cutting events are independent events. If you count fragments rather than cutting events then imho you throw away half of the observations and notably reduce power for differential analysis.
Hi ATpoint! I have a follow up question for this. Would you shift the reads from the bam files before featureCounts, or would you just input exactly the same bam files that you used for peak calling? My concern comes from the way peaks are called by Genrich in ATAC-seq mode, with peaks being called from intervals centered on Tn5 cut sited (end of reads). Shouldn't then the bam files reads be centered to the cut sites as well to achieve a more accurate read count within the peak ranges, or this doesn't matter at all?
I had already opened another thread for asking this before I came here (https://www.biostars.org/p/448204/). I hope that it's fine that I ask here as well, but I would really like to have your opinion on this topic
Hello, the paper below introduce main ATAC-seq analysis steps and many software/packages. I think is useful for beginner.
Yan, F., Powell, D.R., Curtis, D.J. et al. From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol 21, 22 (2020). https://doi.org/10.1186/s13059-020-1929-3
The csaw package does its own peak calling before allowing you to do the differential binding/accessibility assessment using the information from the replicates.




Thanks ATpoint, would it be different for biological replicates rather than technical? I have three biological replicates per condition.
I was indeed referring to biological replicates.