Entering edit mode

4.8 years ago
Anand Rao
▴
640
Greetings!
I want to check whether my BAM file suffers from GC-bias, especially because this is a relatively very old sample, with the following details:
- Tissue Extraction Kit = GeneAll HybridR+ kit (GeneAll)
- RNA Extraction Kit = TruSeq RNA sample preparation kit
- Platform = Illumina HiSeq 2000
- Sequencing Kit = TruSeq SBS kit v3-HS
Based on several posts here on BioStars and elsewhere online, I chose to use
- ComputeGCBias from deepTools, and
- CorrectGCBias, also from deepTools.
I've generated plots for 4 different combos:
- before vs after correcting for GC-bias,
- using either the full unaltered genome assembly or the repeat masked genome assembly.
I seek your help with making sure my interpretation of these results are correct, so here is a composite image of the plots.
If you can confirm / refute my statements, and answer my questions, with any relevant links and commentary, I'd be most obliged. Thanks in advance!
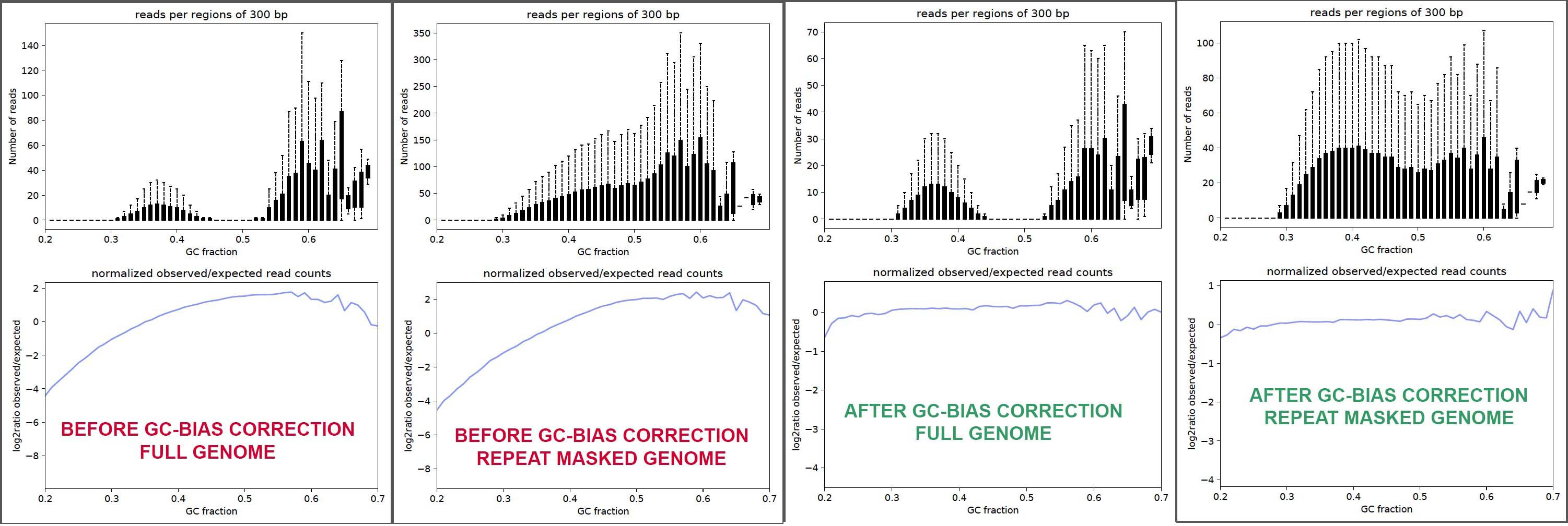
- There is GC-bias in my input sample (red font marked plots), as seen in the bottom sub-panels, whether it be using the full genome or the repeat-masked genome.
- GC-bias correction works as expected in terms of smoothing log2(observed/expected) Vs GC% in the corrected output (green font plots)
- Post-GCbias correction, it looks like for full genome case, reads in the >60% GC-range have been down-sampled.
- I suppose it does not make sense to examine reads against the masked version of the genome because of the possibility that expression can truly be from repeat regions, and other reason(s) ?
- Should I use GC-bias corrected BAM file with featureCounts to report gene expression as count data?
- OR Is this step ONLY to examine and acknowledge if there is GC-bias, but use other preferred methods to compensate for GC-bias?
- If latter, then which bioinformatic protocol(s) to follow for factoring in GC-bias at a later stage in my analyses?


Indeed, I had not realized that Benjamini & Speed algorithm applies only to DNA-Seq. Thanks for pointing that out and for your recommendation for
salmonusing the--gcBiasflag.So this means computeGCBias itself is not valid for RNA-Seq data, let alone correctGCBias - am I right?
Edit: Salmon requires paired-end data.
I guess not, though I am not at all an expert in that matter. The DNA-seq method (from what I take from the video) uses a sliding window approach (probably across the genome), but as in RNA-seq we have coverage mostly in exons, few or absent in introns and essentially no coverage in intergenic regions so this probably messes up the assumptions of that tool. I guess currently salmon is the go-to for this task in RNA-seq. In any case the salmon selective alignment approach typically outperforms traditional alignment for RNA-seq quantification, check the recent benchmarking papers towards that.
From this
ALPINElink, under Description:My data is un-stranded BUT single end RNA-Seq data - so it looks like I cannot use
ALPINE? Is it because using SE data would violate the underlying (statistical) assumptions of PE data, just curious...About SALMON compatibilty with SE vs PE Illumina data, I found this useful excerpt from SALMON README online,
So using SALMON should not be a problem for my SE data.
However, for visualization and verification whether GC-bias exists in SE RNA-Seq data, can I use
SALMONinstead ofALPINE? Or is bias visualization possible only viaALPINE? But since it won't work for my SE data, what tool(s) could I use to visualize bias in my SE RNA-Seq data? Any advice?PS. Just emailed Prof. Michael Love, his inputs should be decisive. He may answer here, or I'll share contents of his reply if I receive one.
I am not aware that it was for unstranded data, the salmon manual does not mention this explicitely but I pinged the developer Rob Patro in our Slack, lets see what he says. Yes PE is necessary. By the way, please consider not to email developers (as many of them explicitely request) and rather open a question at
support.bioconductor.orgwhere many of the Bioc developers including ML are very active and helpful. It is also not standard to correct GC bias, many pipelines do not do this, and if you do not have PE data then why not considering to simply not do it?Thanks for that suggestion, I'd never posted there before, but I just did. Hopefully it's not flagged for cross-posting.
You said
I'm really confused now, because at this SALMON link, it clearly says otherwise:
Could you please clarify with author Rob Patro? Sorry for the bother...
For salmon quantification in general: single- and paired-end data are ok.
For GC-bias correction with salmon: only paired-end data are supported, but it does not matter whether data are stranded or not.
OK, I see what you mean - SE OK for SALMON based quantification, but not for correction. Are there any tools that can visualize and/or correct GC-bias in SE data? May be not, which is probably why you said earlier -