Entering edit mode

4.9 years ago
alyyha
•
0
Hi! I have 2 samples swapped in a PCA of mRNA seq data of 4 genotypes and 3 replicates each: Col0.1 and tbl29.1.
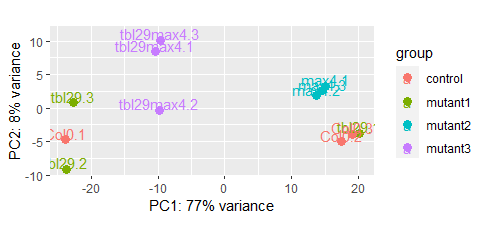
I tried to use VCF files to look for variants, but I do not see any variation. The mutation is an insertion of 639 bp. I tried IGV with bam files (and also VCF) but I do not see huge differences in alignments. With DESeq2 I saw the mutated gene (tbl29) is not expressed in all the replicates (and it´s ok, it can happen, but then I don´t know how to use these mRNA data for determining the swapped samples)
I am getting a bit crazy to demonstrate, on a genotype analysis, that I swapped the 2 samples. Perhaps someone already experienced that and can give me suggestions? I am sure I am doing a very stupid mistake. Thank you very much!
Do you have library or cDNA left to do PCR-based confirmation given that the mutation can be visualized using PCR bands? I mean, the clustering is pretty clear it seems? Maybe just accept it and relabel the samples? You can additionally do something like a Pearson correlation matrix with
cor()for additional confirmation.Unluckily I do not have cDNA for all samples (used almost all) and I cannot do a PCR to show that. I know it looks clear as you said, but I would need to demonstrate it in a way that is undiscussable, that is a sample swap and nothing else.... A Pearson correlation between replicates would group the replicates showing that those 2 are swapped? Or perhaps I could do it using other data like multiple VCF files? For this PCA I used normalized read counts from DESeq2
Yes, biological replicates should have highest correlation. You can additionally do a heatmap and cluster the sample-wise distances as described in the DESeq2 vignette. Then you have three independent computational approaches.
Ok, I will follow your suggestions. Thank you very much!!