Entering edit mode

4.1 years ago
Mat
▴
80
I found a quite big difference in the duplication between Fastp and Fastqc. For all my ~40 SE RNAseq samples, the rate is around 10-30% lower in Fastp compared to Fastqc. Is there an explanation for this and which one should trust more for RNAseq data?
In this scatterplot, the duplication rates for both tools were calculated based on the raw (i.e. untrimmed) reads.
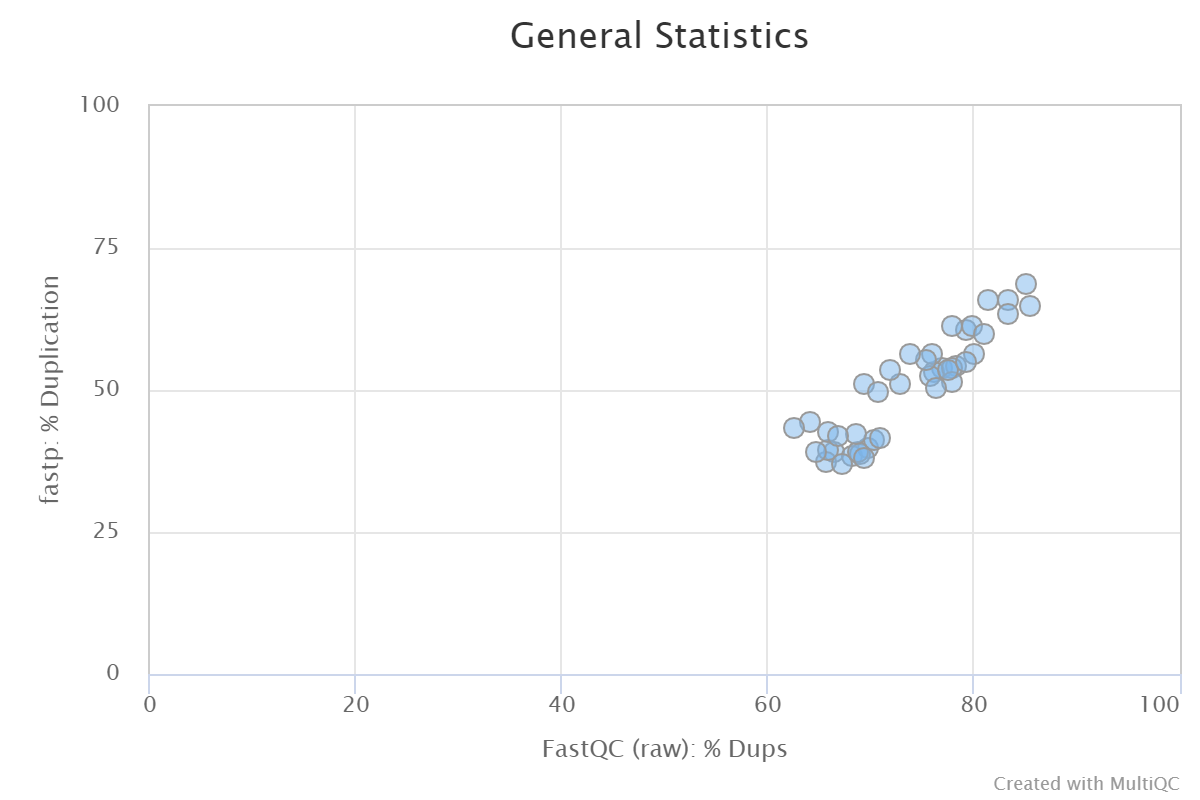
I posted this originally on Github.
You would expect to see duplication in RNAseq of any kind. This is because there are multiple copies of RNA from many genes in your samples. Why are you concerned about this?
Did you even read my question? ;-) It's not about the duplication rate in general, but the huge difference between Fastqc and Fastp.
Fastqc does not look at your entire dataset (I don't know about
fastp) when it checks read duplication. It only uses sequences which first appear in the first 100,000 sequences in each file for this module. While this is generally representative of data it looks like they may not be in your case (especially if fastp is looking at either entire data or some other subset of the data). Most of the QC results for FastQC are representative of the overall data and generally work well.If you truly want to identify sequence duplicates in your data I would recommend using
clumpify.shfrom BBMap suite. A: Introducing Clumpify: Create 30% Smaller, Faster Gzipped Fastq Files Use the optionaddcounts=tto get sequence duplication counts for each sequence type.