Entering edit mode

5.0 years ago
sidwell
▴
10
I use RSEM to count abundance of each transcript after alignment with STAR and featureCounts to count abundance of each transcript after alignment with Bowtie.
My question is, if a read map on a given location of the genome, but at this location, there are multiple transcripts overlapping : How RSEM or featureCounts decide which transcript is it ?
For example, in the mouse at this location : https://www.ensembl.org/Mus_musculus/Location/View?r=7:44804176-44853021;db=core;g=ENSMUSG00000074141
You can see il4i1 and nup62 are overlapping.
But in my counts table, I have a big difference between abundances of those 2 genes. Why ?


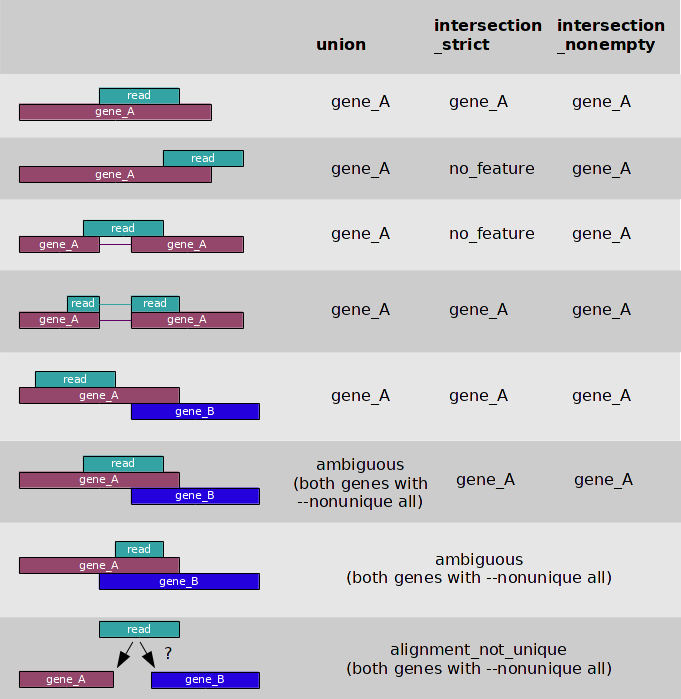

I thought multi-mapping reads was about a read which is mapping at multiple locations. But here my read map at ONE position. But this position is corresponding to multiple transcripts.
The same applies to reads assigned to overlapping features: featureCounts discards them, RSEM tries to optimally assign the counts.