I think that it can be useful to learn to plot this using R, from code. there is a crucial sort of "realization" i had with this is that you create a "cumulative base pair" transformation, so if all your chromosomes are 1000bp long, then you make chr1 occupy the number space 1-1000, then then chr2 is 1001-2000, chr3 from 2001-3000. there are some nice one liners in R that do this, and afterwards, you can tell R to draw the chromosome labels at these given coordinates.
I created simulated reads for yeast, then calculated coverage with mosdepth (https://github.com/brentp/mosdepth), and then loaded the resulting coverage into R with simple read.table commands and plotted with ggplot2
Here is the "setup"
Create a "quickalign" script that aligns paired end reads and outputs mosdepth coverage
the quickalign script
#!/bin/bash
samtools faidx $1
minimap2 -t 8 -a -x sr "$1" "$2" "$3" | \
samtools fixmate -u -m - - | \
samtools sort -u -@2 - | \
samtools markdup -@8 --reference "$1" - --write-index "$4"
mosdepth $4 -b 100 -f $1 $4
Run quickalign over some simulated reads, generated by wgsim
wgsim GCF_000146045.2_R64_genomic.fna a1.fq a2.fq
wgsim GCF_000146045.2_R64_genomic.fna b1.fq b2.fq
wgsim GCF_000146045.2_R64_genomic.fna c1.fq c2.fq
quickalign_paired.sh GCF_000146045.2_R64_genomic.fna a1.fq a2.fq out1.cram
quickalign_paired.sh GCF_000146045.2_R64_genomic.fna b1.fq b2.fq out2.cram
quickalign_paired.sh GCF_000146045.2_R64_genomic.fna c1.fq c2.fq out3.cram
R session for plotting coverage
library(tidyverse)
x1=read.table('out1.cram.regions.bed.gz')
x2=read.table('out2.cram.regions.bed.gz')
x3=read.table('out3.cram.regions.bed.gz')
colnames(x1)<-c("chr","start","end","score")
colnames(x2)<-c("chr","start","end","score")
colnames(x3)<-c("chr","start","end","score")
chrom_sizes=read.table('GCF_000146045.2_R64_genomic.fna.fai')
colnames(chrom_sizes)<-c("chr","size")
chrom_sizes=chrom_sizes[,c(1,2)]
data_cum <- chrom_sizes %>%
group_by(chr) %>%
summarise(max_bp = max(size)) %>%
arrange(-max_bp) %>%
mutate(bp_add = lag(cumsum(as.numeric(max_bp)), default = 0))
process <- function(df) {
df %>%
inner_join(data_cum, by = "chr") %>%
mutate(bp_start_cum = start + bp_add) %>%
mutate(bp_end_cum = end + bp_add)
}
data1 <- process(x1)
data2 <- process(x2)
data3 <- process(x3)
data1$id <- "id1"
data2$id <- "id2"
data3$id <- "id3"
df <- rbind(data1, data2, data3)
axis_set <- data_cum %>%
group_by(chr) %>%
mutate(center = bp_add)
p<-ggplot(data = df) +
geom_point(aes(x=bp_start_cum, y=score),size=0.1,alpha=0.2) +
scale_x_continuous(label = axis_set$chr, breaks = axis_set$center) +
facet_grid(rows = vars(id)) +
theme(
axis.text.x = element_text(angle = 60, size = 8, vjust = 0.5),
axis.title.x=element_blank()
)
ggsave('out.png',width=10,height=4)

Note: I adapted some code I wrote for this answer How to create a graph that shows pairwise Fst values at single SNP loci across the all chromosomes in R

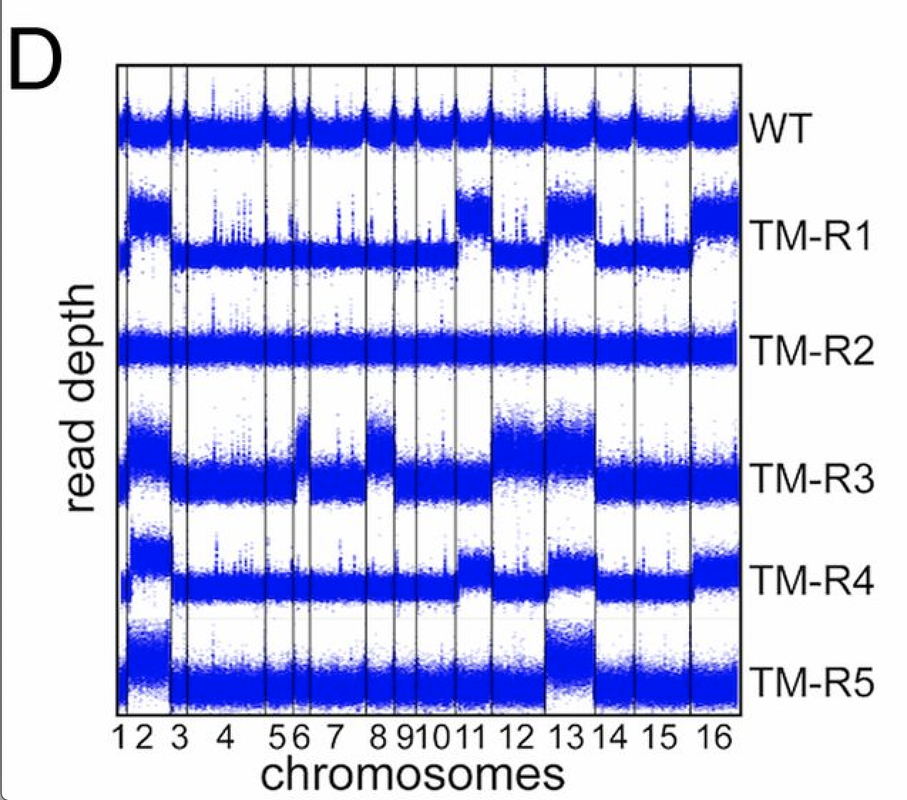




tbh, it looks like a CNV caller worked here =) the plots looks too uniform - which means they were likely GC/library size normalized before plotting
or it is a rolling median across the coverage precalculated in some windows (e.g. 50kbps)
yes, This is true, but it doesn't really answer the question. The question was how this plot was done. can some please help me too?
You can calculate the coverage per some window, smooth it with rolling median and plot in R - this is what I would do