
We have a mutant (gene KO) that displays two different phenotypes identified by the presence or absence of expression of a reporter. Since this mutation is in gene that is part of an endogenous RNAi pathway, and the strains have the same genotype, we hypothesized that the report status is not due to the mutation but "randomly", and that other genes will be affected in the same way: up or down-regulated independently for each mutant strain individual.
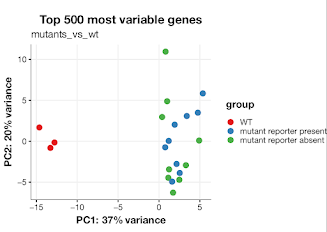
Starting out with gene counts and DESeq2 (experimental set-up bellow), I did some standard clustering (PCA / heatmap of most variable genes) which seems to bore out our hypothesis: replicates within sample cluster together, the wild-types are an out group, but the mutant strains don't show any particular clustering pattern. I am trying to find DEG in each strain vs WT to test if there is any group of DEG which are stable across the strains, and if there is any difference between reporter status.
Questions
Is this the best way to go about this? What other alternatives can I follow to test independent of gene expression from genoype? Very crudely what I want to test is gene expression changes are by and large stochastic for this particular mutant.
Experimental set-up
- WT, 3 replicates
- 3 mutant isolates with phenotype 1 (reporter presence), each in triplicate
- 3 mutant isolates with phenotype 2 (reporter absence), each in triplicate
- each mutant isolate comes from a single individual animal
- poly-A RNA-seq