Edit: the information in this answer has been supplemented (and superseded) by what I have described in this comment in the comments section below..
So I managed to write something in R that approximates this particular alignment plot from the OP given a MSA as an input.
The function (christened msavis()) can be found at my GitHub repo here. Refer to the in-function documentation + comments for details.
But in brief, align your sequences beforehand with your favorite alignment tool (e.g., Mafft or Clustal Omega), and save it as a fasta file. Then invoke msavis() by providing it the path to this fasta file along with the name of a "reference" sequence (can be any one of the sequences in the MSA; e.g., Ref0 in the example below), and an indication of whether the reference sequence should be placed at the top (refontop = TRUE) or at the bottom (refontop = FALSE).
So if you were to test this function with the following toy MSA:
>Ref0
TTGCTACACAACAGCAACAGTCACAGACATTGTCAGTAAC---GCAGCAGCAGCAGCAGCAGAGCTCCCAGG
AGCAGCAGCTCACTTCAGTTCAGCAACCATCTCAGGCTCAGCTGACCCAGCCACCGCAACAATTTTTACAGA
CTTCTAGGTTGCTCCATGGGAATCCCTCAACTCAACTCATTCTCTCTGCTGCATTTCCTCTACAACAGAGCA
CCTTCCCTCAGTCACATCACCAGCAACATCAGTCT
>Seq1
TTGCTACACAACAGCAACAGTCACAGACATTGTCAGTAAC---TCAGCAGCGGCAGCAGCAGAGCTCCCAGG
AGCAGCAGCTCACTTCAGTTCAGCAACCATCTCAGGCTCAGCTGACCCAGCCACCGCAACAATTTTTACAGA
CTTCTAGGTTGCTCCATGGGAATCCCTCAACTCAACTCATTCTCTCTGCTGCATTTCCTCTACAACAGAGCA
CCTTCCCTCAGTCACATCACCAGCAACATCAGTCT
>Seq2
TTGCTACACAACAG---CAGTCACAGACATTGTCAGTCACACAGCAGCAGCAGCAGCAGCAGAGTGGCCAGG
AACAGCAGCTTGCTTCAGTTCAGCAACCGTCTCAGGCTCAGCTGACCCAACCACCACAGCAGTTTTTACAGA
CTTCTAGGTTGCTCCATGGGAATCCTTCAACTCAGCTTATCCTCTCTGCTGCATTTCCACTACAACAGAGCA
CTTTCCCTCAATCACATCACCAGCAGCACCAGTCT
>Seq3
TTGCTCCACAACAACCGCAGTCGCAGACACTGTCAGTCAC---------GCAGCAGCAGCAGAGTTCCCAGG
AACAGCCGCTCCCTTCAGTTCAGCAACCATCTCAGGCTCAGCTGACTCAACCACCGCAGCAGTTTCTACAGA
CTTCTAGGTTGCTCCATGGGAACCCTTCAACTCAGCTTATCCTCTCTGCTGCATTTCCTCTACAGCAGAGCA
CTTTCCCTCAGTCACATCACCAGCAACACCAGTCG
>Seq4
TTGCTCCACAACAACCGCAGTCACAGACACTGTCAGTCAC---------GCAGCAGCAGCAGAGTTCCCAGG
AACAGCCTCTCCCTTCAGTTCAGCAACCATCTCAGGCTCAGCTGACTCAACCACCGCAGCAGTTTTTACAGA
CTTCCAGGTTGCTCCATGGGAACCCTTCAACTCAGCTTATCCTCTCTGCTGCATTTCCTCTACAGCAGAGCA
CTTTCCCTCAGTCACATCACCAGCAACACCAGTCG
>Seq5
TTGCCACACAACAGCAACAGCAGCAGCAACAGCAGCAGCA---GCAGCAGCAACAGCAGCAGAGCTCCCAAG
AGCAGCAGCTTCCTGCAGTTCAGCAACCATCTCAGGCCCAGCTGACCCAGCCACCACAGCAGTTTTTACAGA
CACCTAGGTTGCTCCATGGGAATCCTTCCGCTCAGCTCATCCTCTCTGCTGCCTTTCCTCTACAACAGAGCA
CCTTCCCTCCGTCGCATCACCAGCAACACCAGCCT
>Seq6
TTGCCACACAACAGCAACAGCAGCAGCAACAGCAGCAGCA---GCAGCAGCAACAGCAGCAGAGCTCCCAAG
AGCAGCAGCTTCCTGCAGTTCAGCAACCATCTCAGGCCCAGCTGACCCAGCCACCACAGCAGTTTTTACAGA
CACCTAGGTTGCTCCATGGGAATCCTTCCGCTCAGCTCATCCTCTCTGCTGCCTTTCCTCTACAACAGAGCA
CCTTTCCTCCGTCGCATCACCAGCAACACCAGCCT
By invoking msavis() like so:
msavis("/path/to/test/alignment.fasta", "Ref0", refontop = TRUE)
You'd get the following plot:
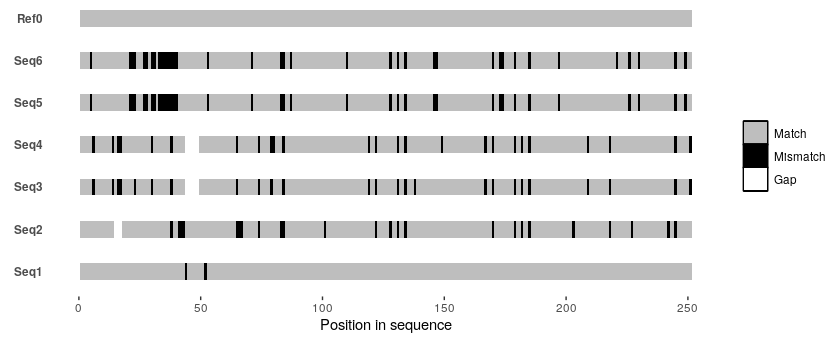
There's probably a lot of additional functionality that can be added to this function, but this is a start. I'm plenty sure there are a couple of bugs in there, so I'd appreciate any and all feedback.



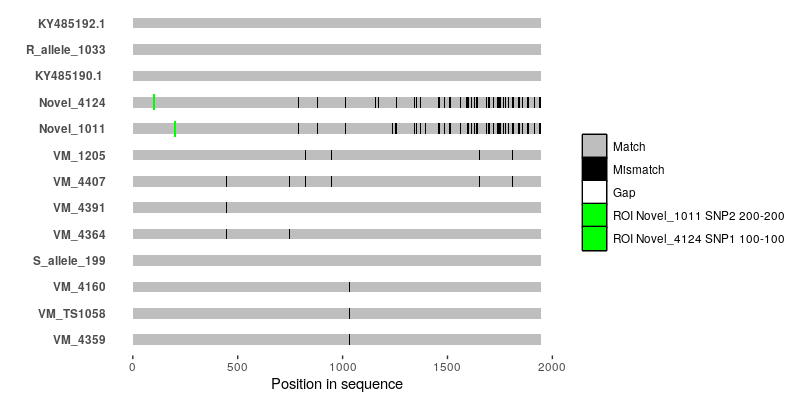
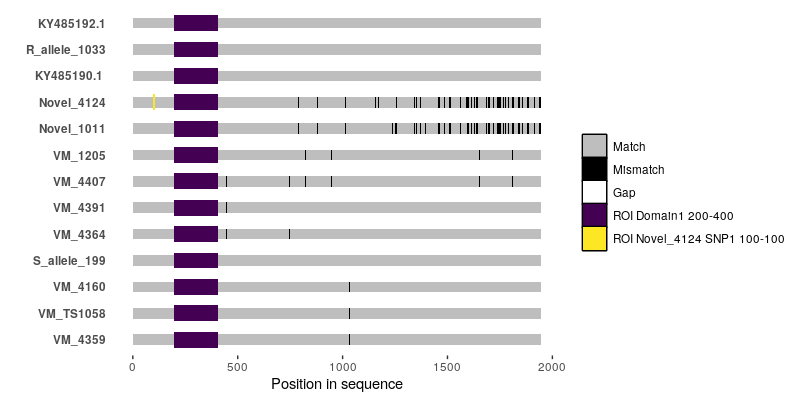


It's probably doable using
ggmsaor by writing something based off ofggplot2from scratch, but you're going to have to tell us what--for instance--the black bars and the white bars in your alignment figure are.So basically, the white bars indicate gaps in the alignment. The black bars are single nucleotide polymorphisms. I checked with the authors of that paper and they told they used a script that they wrote for this graph but they lost the script. They also did not mention which software they used. That is why I am really confused.