Entering edit mode

4.0 years ago
rthapa
▴
90
Hi,
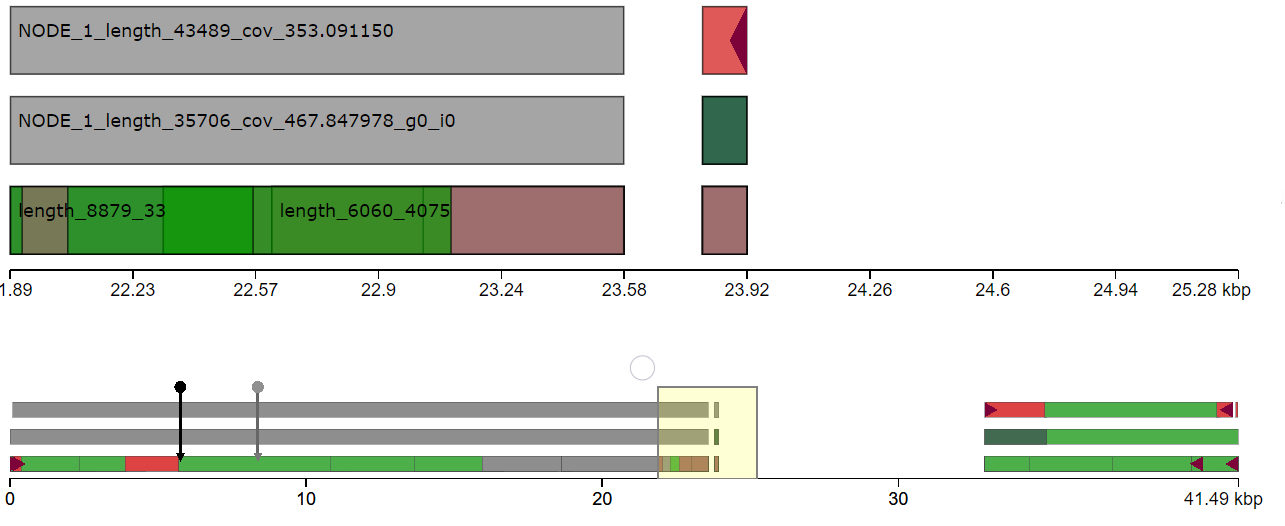
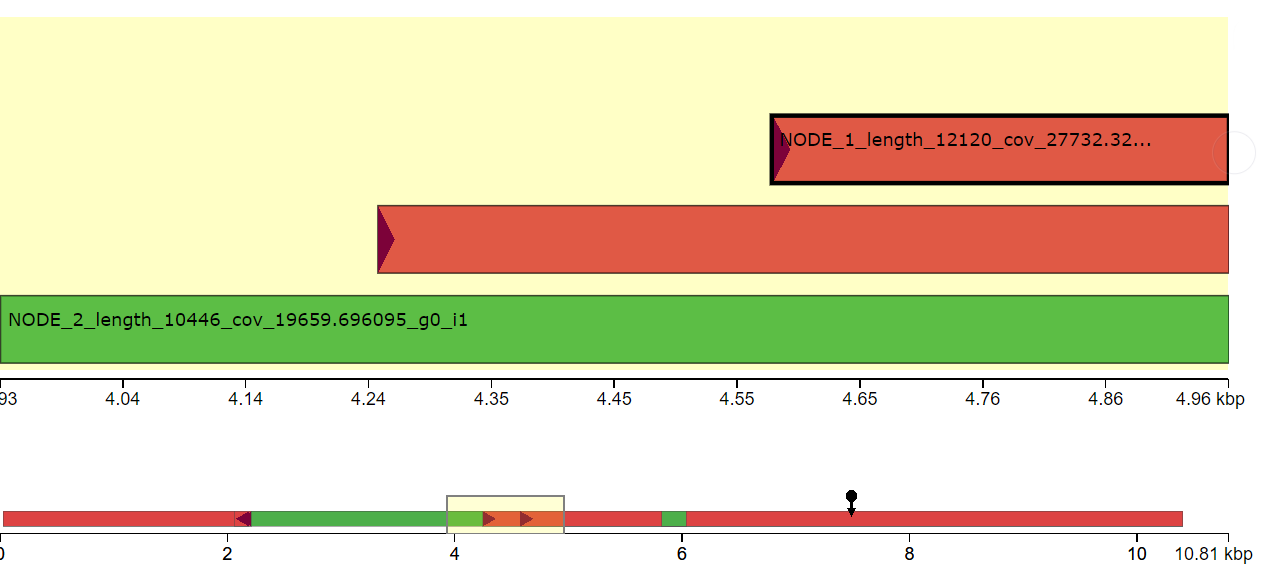
I have done de novo genome assembly of a bacterial strain using canu. I want to find structural variants comparing with the reference genome. Since, the bacterial genome is circular, It is hard to find the origin of replication to align with the reference genome. Does anyone have suggestions how to find structural variants in the bacterial genome comparing with the reference genome?
Thanks


This question has been asked a couple of different ways: de novo genome assembly of bacterial genome
rthapa : Have you tried to repeat the assembly? Perhaps you will get an assembly that will be co-linear with the reference.
Yes, I tried to repeat the assembly after removing read lengths shorter than 2000 bp. The assembly result is similar. I think the issue is due to incorrect identification of origin of replication during genome assembly.
Where is the
dnaAgene located in your assembly and where is it in the reference? Paper1 and Paper2.