Entering edit mode

3.9 years ago
Matt
▴
20
Hi,
I would like to extract the 2 biological reads of a RNAseq single cell of a paired-end sequencing.
With this run SRR11772847 I tried the command line of the sra-toolkit ./fastq-dump --skip-technical --split-3 SRR11772847
I should have 2 .fastq but I only get one with reads of size 98 bp (there is an extract below), there are 496352056 lines
@SRR11772847.1.3 NB502129:188:HY73HBGX9:1:11101:13593:1050 length=98
NCGATCCNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+SRR11772847.1.3 NB502129:188:HY73HBGX9:1:11101:13593:1050 length=98
#AAAAEA###########################################################################################
@SRR11772847.2.3 NB502129:188:HY73HBGX9:1:11101:9270:1050 length=98
NCCATGTNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+SRR11772847.2.3 NB502129:188:HY73HBGX9:1:11101:9270:1050 length=98
#A/AA//###########################################################################################
@SRR11772847.3.3 NB502129:188:HY73HBGX9:1:11101:16784:1053 length=98
NAAAAGAATATCTGTCCTANNGNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+SRR11772847.3.3 NB502129:188:HY73HBGX9:1:11101:16784:1053 length=98
#A/AAEEEEEEAEEEEEAA##E############################################################################
@SRR11772847.4.3 NB502129:188:HY73HBGX9:1:11101:20118:1053 length=98
NAGGAGGATGAAGGCTTACNNGNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+SRR11772847.4.3 NB502129:188:HY73HBGX9:1:11101:20118:1053 length=98
#A6AAE/AE/E/E/EEA//##<############################################################################
@SRR11772847.5.3 NB502129:188:HY73HBGX9:1:11101:13559:1054 length=98
NTTTTAGTTGGTCTTCATCTNTNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+SRR11772847.5.3 NB502129:188:HY73HBGX9:1:11101:13559:1054 length=98
#AAAA/<E//EEEE6A/AE/#<############################################################################
I'm quite a beginner, do I miss something or the data for this run is incomplete ? I have an analogical problem with SRR7049900 run
i also tried ./fastq-dump -I --split-files SRR11772847 but get 3 fastq with reads of size 8bp, 26bp and 98bp. I should get an other fastq of size 98bp (read2 of the paired-end sequencing), i don't understand.
Thank you by advance for your help,
Matt

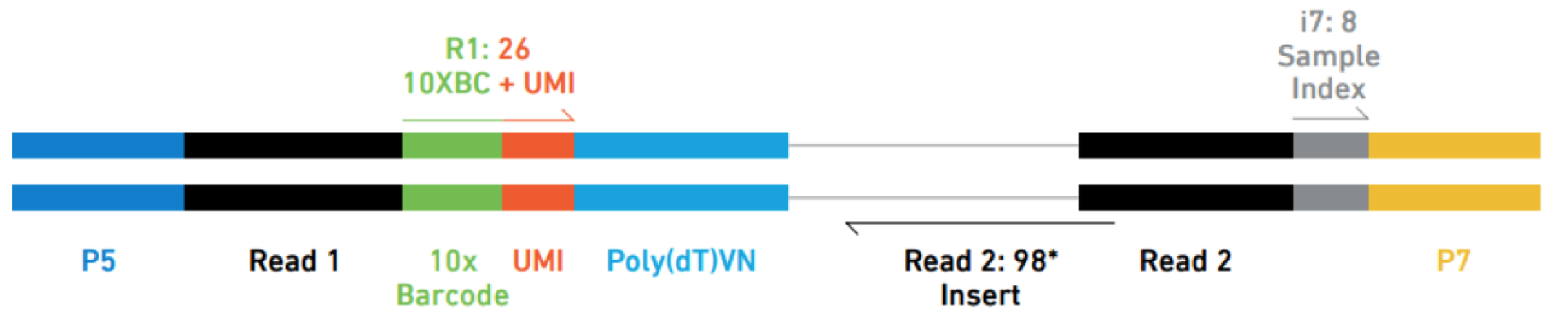
Thank you very much for this answer, very clear very complete, i successfully used Cellranger with that.