Entering edit mode

3.8 years ago
storm1907
▴
30
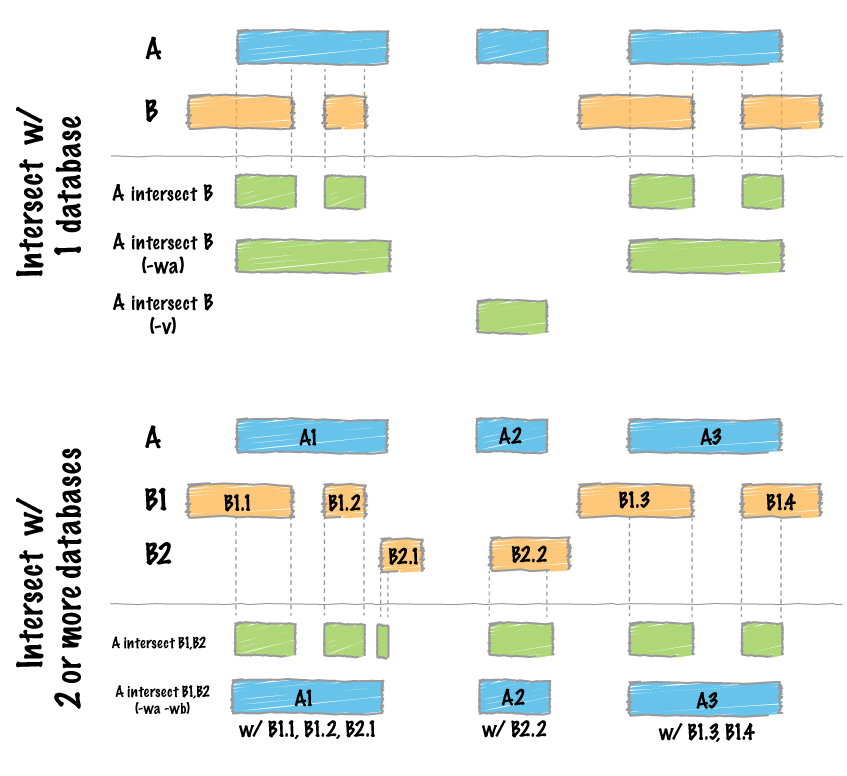
Hello, I am trying to compare genome vcf file with bed file, to get out from genome file only these positions present in bed file
which syntax could be correct: bedtools intersect -a file.vcf -b file1.bed or bedtools intersect -a file1.bed -b file.vcf ?
I am confused, and also second variant seems to be correct, but is not working

Thank you! But now I got this:
this is not VCF file or a BED file. Show us the header of both files.