|
# EXAMPLE USAGE |
|
|
|
# example of colsidecolors rowsidecolors (single column, single row) |
|
mat <- matrix(1:100, byrow=T, nrow=10) |
|
column_annotation <- sample(c("red", "blue", "green"), 10, replace=T) |
|
column_annotation <- as.matrix(column_annotation) |
|
colnames(column_annotation) <- c("Variable X") |
|
|
|
row_annotation <- sample(c("red", "blue", "green"), 10, replace=T) |
|
row_annotation <- as.matrix(t(row_annotation)) |
|
rownames(row_annotation) <- c("Variable Y") |
|
|
|
heatmap.3(mat, RowSideColors=row_annotation, ColSideColors=column_annotation) |
|
|
|
# multiple column and row |
|
mat <- matrix(1:100, byrow=T, nrow=10) |
|
column_annotation <- matrix(sample(c("red", "blue", "green"), 20, replace=T), ncol=2) |
|
colnames(column_annotation) <- c("Variable X1", "Variable X2") |
|
|
|
row_annotation <- matrix(sample(c("red", "blue", "green"), 20, replace=T), nrow=2) |
|
rownames(row_annotation) <- c("Variable Y1", "Variable Y2") |
|
|
|
heatmap.3(mat, RowSideColors=row_annotation, ColSideColors=column_annotation) |
|
|
|
|
|
# CODE |
|
|
|
heatmap.3 <- function(x, |
|
Rowv = TRUE, Colv = if (symm) "Rowv" else TRUE, |
|
distfun = dist, |
|
hclustfun = hclust, |
|
dendrogram = c("both","row", "column", "none"), |
|
symm = FALSE, |
|
scale = c("none","row", "column"), |
|
na.rm = TRUE, |
|
revC = identical(Colv,"Rowv"), |
|
add.expr, |
|
breaks, |
|
symbreaks = max(x < 0, na.rm = TRUE) || scale != "none", |
|
col = "heat.colors", |
|
colsep, |
|
rowsep, |
|
sepcolor = "white", |
|
sepwidth = c(0.05, 0.05), |
|
cellnote, |
|
notecex = 1, |
|
notecol = "cyan", |
|
na.color = par("bg"), |
|
trace = c("none", "column","row", "both"), |
|
tracecol = "cyan", |
|
hline = median(breaks), |
|
vline = median(breaks), |
|
linecol = tracecol, |
|
margins = c(5,5), |
|
ColSideColors, |
|
RowSideColors, |
|
side.height.fraction=0.3, |
|
cexRow = 0.2 + 1/log10(nr), |
|
cexCol = 0.2 + 1/log10(nc), |
|
labRow = NULL, |
|
labCol = NULL, |
|
key = TRUE, |
|
keysize = 1.5, |
|
density.info = c("none", "histogram", "density"), |
|
denscol = tracecol, |
|
symkey = max(x < 0, na.rm = TRUE) || symbreaks, |
|
densadj = 0.25, |
|
main = NULL, |
|
xlab = NULL, |
|
ylab = NULL, |
|
lmat = NULL, |
|
lhei = NULL, |
|
lwid = NULL, |
|
NumColSideColors = 1, |
|
NumRowSideColors = 1, |
|
KeyValueName="Value",...){ |
|
|
|
invalid <- function (x) { |
|
if (missing(x) || is.null(x) || length(x) == 0) |
|
return(TRUE) |
|
if (is.list(x)) |
|
return(all(sapply(x, invalid))) |
|
else if (is.vector(x)) |
|
return(all(is.na(x))) |
|
else return(FALSE) |
|
} |
|
|
|
x <- as.matrix(x) |
|
scale01 <- function(x, low = min(x), high = max(x)) { |
|
x <- (x - low)/(high - low) |
|
x |
|
} |
|
retval <- list() |
|
scale <- if (symm && missing(scale)) |
|
"none" |
|
else match.arg(scale) |
|
dendrogram <- match.arg(dendrogram) |
|
trace <- match.arg(trace) |
|
density.info <- match.arg(density.info) |
|
if (length(col) == 1 && is.character(col)) |
|
col <- get(col, mode = "function") |
|
if (!missing(breaks) && (scale != "none")) |
|
warning("Using scale=\"row\" or scale=\"column\" when breaks are", |
|
"specified can produce unpredictable results.", "Please consider using only one or the other.") |
|
if (is.null(Rowv) || is.na(Rowv)) |
|
Rowv <- FALSE |
|
if (is.null(Colv) || is.na(Colv)) |
|
Colv <- FALSE |
|
else if (Colv == "Rowv" && !isTRUE(Rowv)) |
|
Colv <- FALSE |
|
if (length(di <- dim(x)) != 2 || !is.numeric(x)) |
|
stop("`x' must be a numeric matrix") |
|
nr <- di[1] |
|
nc <- di[2] |
|
if (nr <= 1 || nc <= 1) |
|
stop("`x' must have at least 2 rows and 2 columns") |
|
if (!is.numeric(margins) || length(margins) != 2) |
|
stop("`margins' must be a numeric vector of length 2") |
|
if (missing(cellnote)) |
|
cellnote <- matrix("", ncol = ncol(x), nrow = nrow(x)) |
|
if (!inherits(Rowv, "dendrogram")) { |
|
if (((!isTRUE(Rowv)) || (is.null(Rowv))) && (dendrogram %in% |
|
c("both", "row"))) { |
|
if (is.logical(Colv) && (Colv)) |
|
dendrogram <- "column" |
|
else dedrogram <- "none" |
|
warning("Discrepancy: Rowv is FALSE, while dendrogram is `", |
|
dendrogram, "'. Omitting row dendogram.") |
|
} |
|
} |
|
if (!inherits(Colv, "dendrogram")) { |
|
if (((!isTRUE(Colv)) || (is.null(Colv))) && (dendrogram %in% |
|
c("both", "column"))) { |
|
if (is.logical(Rowv) && (Rowv)) |
|
dendrogram <- "row" |
|
else dendrogram <- "none" |
|
warning("Discrepancy: Colv is FALSE, while dendrogram is `", |
|
dendrogram, "'. Omitting column dendogram.") |
|
} |
|
} |
|
if (inherits(Rowv, "dendrogram")) { |
|
ddr <- Rowv |
|
rowInd <- order.dendrogram(ddr) |
|
} |
|
else if (is.integer(Rowv)) { |
|
hcr <- hclustfun(distfun(x)) |
|
ddr <- as.dendrogram(hcr) |
|
ddr <- reorder(ddr, Rowv) |
|
rowInd <- order.dendrogram(ddr) |
|
if (nr != length(rowInd)) |
|
stop("row dendrogram ordering gave index of wrong length") |
|
} |
|
else if (isTRUE(Rowv)) { |
|
Rowv <- rowMeans(x, na.rm = na.rm) |
|
hcr <- hclustfun(distfun(x)) |
|
ddr <- as.dendrogram(hcr) |
|
ddr <- reorder(ddr, Rowv) |
|
rowInd <- order.dendrogram(ddr) |
|
if (nr != length(rowInd)) |
|
stop("row dendrogram ordering gave index of wrong length") |
|
} |
|
else { |
|
rowInd <- nr:1 |
|
} |
|
if (inherits(Colv, "dendrogram")) { |
|
ddc <- Colv |
|
colInd <- order.dendrogram(ddc) |
|
} |
|
else if (identical(Colv, "Rowv")) { |
|
if (nr != nc) |
|
stop("Colv = \"Rowv\" but nrow(x) != ncol(x)") |
|
if (exists("ddr")) { |
|
ddc <- ddr |
|
colInd <- order.dendrogram(ddc) |
|
} |
|
else colInd <- rowInd |
|
} |
|
else if (is.integer(Colv)) { |
|
hcc <- hclustfun(distfun(if (symm) |
|
x |
|
else t(x))) |
|
ddc <- as.dendrogram(hcc) |
|
ddc <- reorder(ddc, Colv) |
|
colInd <- order.dendrogram(ddc) |
|
if (nc != length(colInd)) |
|
stop("column dendrogram ordering gave index of wrong length") |
|
} |
|
else if (isTRUE(Colv)) { |
|
Colv <- colMeans(x, na.rm = na.rm) |
|
hcc <- hclustfun(distfun(if (symm) |
|
x |
|
else t(x))) |
|
ddc <- as.dendrogram(hcc) |
|
ddc <- reorder(ddc, Colv) |
|
colInd <- order.dendrogram(ddc) |
|
if (nc != length(colInd)) |
|
stop("column dendrogram ordering gave index of wrong length") |
|
} |
|
else { |
|
colInd <- 1:nc |
|
} |
|
retval$rowInd <- rowInd |
|
retval$colInd <- colInd |
|
retval$call <- match.call() |
|
x <- x[rowInd, colInd] |
|
x.unscaled <- x |
|
cellnote <- cellnote[rowInd, colInd] |
|
if (is.null(labRow)) |
|
labRow <- if (is.null(rownames(x))) |
|
(1:nr)[rowInd] |
|
else rownames(x) |
|
else labRow <- labRow[rowInd] |
|
if (is.null(labCol)) |
|
labCol <- if (is.null(colnames(x))) |
|
(1:nc)[colInd] |
|
else colnames(x) |
|
else labCol <- labCol[colInd] |
|
if (scale == "row") { |
|
retval$rowMeans <- rm <- rowMeans(x, na.rm = na.rm) |
|
x <- sweep(x, 1, rm) |
|
retval$rowSDs <- sx <- apply(x, 1, sd, na.rm = na.rm) |
|
x <- sweep(x, 1, sx, "/") |
|
} |
|
else if (scale == "column") { |
|
retval$colMeans <- rm <- colMeans(x, na.rm = na.rm) |
|
x <- sweep(x, 2, rm) |
|
retval$colSDs <- sx <- apply(x, 2, sd, na.rm = na.rm) |
|
x <- sweep(x, 2, sx, "/") |
|
} |
|
if (missing(breaks) || is.null(breaks) || length(breaks) < 1) { |
|
if (missing(col) || is.function(col)) |
|
breaks <- 16 |
|
else breaks <- length(col) + 1 |
|
} |
|
if (length(breaks) == 1) { |
|
if (!symbreaks) |
|
breaks <- seq(min(x, na.rm = na.rm), max(x, na.rm = na.rm), |
|
length = breaks) |
|
else { |
|
extreme <- max(abs(x), na.rm = TRUE) |
|
breaks <- seq(-extreme, extreme, length = breaks) |
|
} |
|
} |
|
nbr <- length(breaks) |
|
ncol <- length(breaks) - 1 |
|
if (class(col) == "function") |
|
col <- col(ncol) |
|
min.breaks <- min(breaks) |
|
max.breaks <- max(breaks) |
|
x[x < min.breaks] <- min.breaks |
|
x[x > max.breaks] <- max.breaks |
|
if (missing(lhei) || is.null(lhei)) |
|
lhei <- c(keysize, 4) |
|
if (missing(lwid) || is.null(lwid)) |
|
lwid <- c(keysize, 4) |
|
if (missing(lmat) || is.null(lmat)) { |
|
lmat <- rbind(4:3, 2:1) |
|
|
|
if (!missing(ColSideColors)) { |
|
#if (!is.matrix(ColSideColors)) |
|
#stop("'ColSideColors' must be a matrix") |
|
if (!is.character(ColSideColors) || nrow(ColSideColors) != nc) |
|
stop("'ColSideColors' must be a matrix of nrow(x) rows") |
|
lmat <- rbind(lmat[1, ] + 1, c(NA, 1), lmat[2, ] + 1) |
|
#lhei <- c(lhei[1], 0.2, lhei[2]) |
|
lhei=c(lhei[1], side.height.fraction*NumColSideColors, lhei[2]) |
|
} |
|
|
|
if (!missing(RowSideColors)) { |
|
#if (!is.matrix(RowSideColors)) |
|
#stop("'RowSideColors' must be a matrix") |
|
if (!is.character(RowSideColors) || ncol(RowSideColors) != nr) |
|
stop("'RowSideColors' must be a matrix of ncol(x) columns") |
|
lmat <- cbind(lmat[, 1] + 1, c(rep(NA, nrow(lmat) - 1), 1), lmat[,2] + 1) |
|
#lwid <- c(lwid[1], 0.2, lwid[2]) |
|
lwid <- c(lwid[1], side.height.fraction*NumRowSideColors, lwid[2]) |
|
} |
|
lmat[is.na(lmat)] <- 0 |
|
} |
|
|
|
if (length(lhei) != nrow(lmat)) |
|
stop("lhei must have length = nrow(lmat) = ", nrow(lmat)) |
|
if (length(lwid) != ncol(lmat)) |
|
stop("lwid must have length = ncol(lmat) =", ncol(lmat)) |
|
op <- par(no.readonly = TRUE) |
|
on.exit(par(op)) |
|
|
|
layout(lmat, widths = lwid, heights = lhei, respect = FALSE) |
|
|
|
if (!missing(RowSideColors)) { |
|
if (!is.matrix(RowSideColors)){ |
|
par(mar = c(margins[1], 0, 0, 0.5)) |
|
image(rbind(1:nr), col = RowSideColors[rowInd], axes = FALSE) |
|
} else { |
|
par(mar = c(margins[1], 0, 0, 0.5)) |
|
rsc = t(RowSideColors[,rowInd, drop=F]) |
|
rsc.colors = matrix() |
|
rsc.names = names(table(rsc)) |
|
rsc.i = 1 |
|
for (rsc.name in rsc.names) { |
|
rsc.colors[rsc.i] = rsc.name |
|
rsc[rsc == rsc.name] = rsc.i |
|
rsc.i = rsc.i + 1 |
|
} |
|
rsc = matrix(as.numeric(rsc), nrow = dim(rsc)[1]) |
|
image(t(rsc), col = as.vector(rsc.colors), axes = FALSE) |
|
if (length(colnames(RowSideColors)) > 0) { |
|
axis(1, 0:(dim(rsc)[2] - 1)/(dim(rsc)[2] - 1), colnames(RowSideColors), las = 2, tick = FALSE) |
|
} |
|
} |
|
} |
|
|
|
if (!missing(ColSideColors)) { |
|
|
|
if (!is.matrix(ColSideColors)){ |
|
par(mar = c(0.5, 0, 0, margins[2])) |
|
image(cbind(1:nc), col = ColSideColors[colInd], axes = FALSE) |
|
} else { |
|
par(mar = c(0.5, 0, 0, margins[2])) |
|
csc = ColSideColors[colInd, , drop=F] |
|
csc.colors = matrix() |
|
csc.names = names(table(csc)) |
|
csc.i = 1 |
|
for (csc.name in csc.names) { |
|
csc.colors[csc.i] = csc.name |
|
csc[csc == csc.name] = csc.i |
|
csc.i = csc.i + 1 |
|
} |
|
csc = matrix(as.numeric(csc), nrow = dim(csc)[1]) |
|
image(csc, col = as.vector(csc.colors), axes = FALSE) |
|
if (length(colnames(ColSideColors)) > 0) { |
|
axis(2, 0:(dim(csc)[2] - 1)/max(1,(dim(csc)[2] - 1)), colnames(ColSideColors), las = 2, tick = FALSE) |
|
} |
|
} |
|
} |
|
|
|
par(mar = c(margins[1], 0, 0, margins[2])) |
|
x <- t(x) |
|
cellnote <- t(cellnote) |
|
if (revC) { |
|
iy <- nr:1 |
|
if (exists("ddr")) |
|
ddr <- rev(ddr) |
|
x <- x[, iy] |
|
cellnote <- cellnote[, iy] |
|
} |
|
else iy <- 1:nr |
|
image(1:nc, 1:nr, x, xlim = 0.5 + c(0, nc), ylim = 0.5 + c(0, nr), axes = FALSE, xlab = "", ylab = "", col = col, breaks = breaks, ...) |
|
retval$carpet <- x |
|
if (exists("ddr")) |
|
retval$rowDendrogram <- ddr |
|
if (exists("ddc")) |
|
retval$colDendrogram <- ddc |
|
retval$breaks <- breaks |
|
retval$col <- col |
|
if (!invalid(na.color) & any(is.na(x))) { # load library(gplots) |
|
mmat <- ifelse(is.na(x), 1, NA) |
|
image(1:nc, 1:nr, mmat, axes = FALSE, xlab = "", ylab = "", |
|
col = na.color, add = TRUE) |
|
} |
|
axis(1, 1:nc, labels = labCol, las = 2, line = -0.5, tick = 0, |
|
cex.axis = cexCol) |
|
if (!is.null(xlab)) |
|
mtext(xlab, side = 1, line = margins[1] - 1.25) |
|
axis(4, iy, labels = labRow, las = 2, line = -0.5, tick = 0, |
|
cex.axis = cexRow) |
|
if (!is.null(ylab)) |
|
mtext(ylab, side = 4, line = margins[2] - 1.25) |
|
if (!missing(add.expr)) |
|
eval(substitute(add.expr)) |
|
if (!missing(colsep)) |
|
for (csep in colsep) rect(xleft = csep + 0.5, ybottom = rep(0, length(csep)), xright = csep + 0.5 + sepwidth[1], ytop = rep(ncol(x) + 1, csep), lty = 1, lwd = 1, col = sepcolor, border = sepcolor) |
|
if (!missing(rowsep)) |
|
for (rsep in rowsep) rect(xleft = 0, ybottom = (ncol(x) + 1 - rsep) - 0.5, xright = nrow(x) + 1, ytop = (ncol(x) + 1 - rsep) - 0.5 - sepwidth[2], lty = 1, lwd = 1, col = sepcolor, border = sepcolor) |
|
min.scale <- min(breaks) |
|
max.scale <- max(breaks) |
|
x.scaled <- scale01(t(x), min.scale, max.scale) |
|
if (trace %in% c("both", "column")) { |
|
retval$vline <- vline |
|
vline.vals <- scale01(vline, min.scale, max.scale) |
|
for (i in colInd) { |
|
if (!is.null(vline)) { |
|
abline(v = i - 0.5 + vline.vals, col = linecol, |
|
lty = 2) |
|
} |
|
xv <- rep(i, nrow(x.scaled)) + x.scaled[, i] - 0.5 |
|
xv <- c(xv[1], xv) |
|
yv <- 1:length(xv) - 0.5 |
|
lines(x = xv, y = yv, lwd = 1, col = tracecol, type = "s") |
|
} |
|
} |
|
if (trace %in% c("both", "row")) { |
|
retval$hline <- hline |
|
hline.vals <- scale01(hline, min.scale, max.scale) |
|
for (i in rowInd) { |
|
if (!is.null(hline)) { |
|
abline(h = i + hline, col = linecol, lty = 2) |
|
} |
|
yv <- rep(i, ncol(x.scaled)) + x.scaled[i, ] - 0.5 |
|
yv <- rev(c(yv[1], yv)) |
|
xv <- length(yv):1 - 0.5 |
|
lines(x = xv, y = yv, lwd = 1, col = tracecol, type = "s") |
|
} |
|
} |
|
if (!missing(cellnote)) |
|
text(x = c(row(cellnote)), y = c(col(cellnote)), labels = c(cellnote), |
|
col = notecol, cex = notecex) |
|
par(mar = c(margins[1], 0, 0, 0)) |
|
if (dendrogram %in% c("both", "row")) { |
|
plot(ddr, horiz = TRUE, axes = FALSE, yaxs = "i", leaflab = "none") |
|
} |
|
else plot.new() |
|
par(mar = c(0, 0, if (!is.null(main)) 5 else 0, margins[2])) |
|
if (dendrogram %in% c("both", "column")) { |
|
plot(ddc, axes = FALSE, xaxs = "i", leaflab = "none") |
|
} |
|
else plot.new() |
|
if (!is.null(main)) |
|
title(main, cex.main = 1.5 * op[["cex.main"]]) |
|
if (key) { |
|
par(mar = c(5, 4, 2, 1), cex = 0.75) |
|
tmpbreaks <- breaks |
|
if (symkey) { |
|
max.raw <- max(abs(c(x, breaks)), na.rm = TRUE) |
|
min.raw <- -max.raw |
|
tmpbreaks[1] <- -max(abs(x), na.rm = TRUE) |
|
tmpbreaks[length(tmpbreaks)] <- max(abs(x), na.rm = TRUE) |
|
} |
|
else { |
|
min.raw <- min(x, na.rm = TRUE) |
|
max.raw <- max(x, na.rm = TRUE) |
|
} |
|
|
|
z <- seq(min.raw, max.raw, length = length(col)) |
|
image(z = matrix(z, ncol = 1), col = col, breaks = tmpbreaks, |
|
xaxt = "n", yaxt = "n") |
|
par(usr = c(0, 1, 0, 1)) |
|
lv <- pretty(breaks) |
|
xv <- scale01(as.numeric(lv), min.raw, max.raw) |
|
axis(1, at = xv, labels = lv) |
|
if (scale == "row") |
|
mtext(side = 1, "Row Z-Score", line = 2) |
|
else if (scale == "column") |
|
mtext(side = 1, "Column Z-Score", line = 2) |
|
else mtext(side = 1, KeyValueName, line = 2) |
|
if (density.info == "density") { |
|
dens <- density(x, adjust = densadj, na.rm = TRUE) |
|
omit <- dens$x < min(breaks) | dens$x > max(breaks) |
|
dens$x <- dens$x[-omit] |
|
dens$y <- dens$y[-omit] |
|
dens$x <- scale01(dens$x, min.raw, max.raw) |
|
lines(dens$x, dens$y/max(dens$y) * 0.95, col = denscol, |
|
lwd = 1) |
|
axis(2, at = pretty(dens$y)/max(dens$y) * 0.95, pretty(dens$y)) |
|
title("Color Key\nand Density Plot") |
|
par(cex = 0.5) |
|
mtext(side = 2, "Density", line = 2) |
|
} |
|
else if (density.info == "histogram") { |
|
h <- hist(x, plot = FALSE, breaks = breaks) |
|
hx <- scale01(breaks, min.raw, max.raw) |
|
hy <- c(h$counts, h$counts[length(h$counts)]) |
|
lines(hx, hy/max(hy) * 0.95, lwd = 1, type = "s", |
|
col = denscol) |
|
axis(2, at = pretty(hy)/max(hy) * 0.95, pretty(hy)) |
|
title("Color Key\nand Histogram") |
|
par(cex = 0.5) |
|
mtext(side = 2, "Count", line = 2) |
|
} |
|
else title("Color Key") |
|
} |
|
else plot.new() |
|
retval$colorTable <- data.frame(low = retval$breaks[-length(retval$breaks)], |
|
high = retval$breaks[-1], color = retval$col) |
|
invisible(retval) |
|
} |

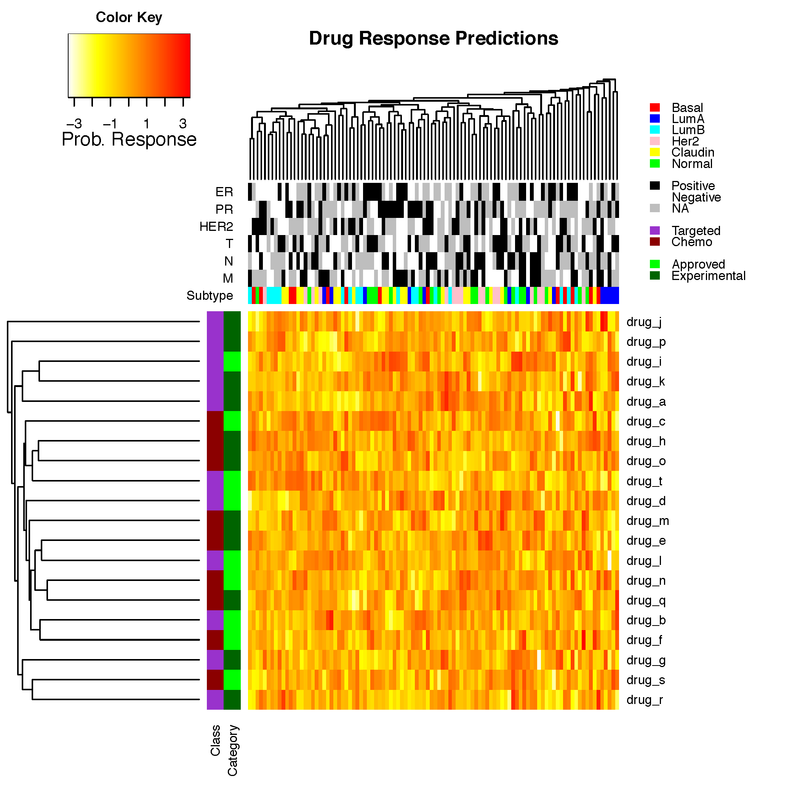




Thanks! That's really useful. BTW, could you provide a complete example to exactly reproduce your figure?
This should have been a comment. Not an answer. I have provided example R code. Unfortunately I am not able to provide a complete example because the data underlying this particular figure is very large and not available for public posting yet.
I found some time and re-worked my answer above to provide a complete working example, now on random data, rather than data I couldn't release here.
cited in : "SEER and Gene Expression Data Analysis Deciphers Racial Disparity Patterns in Prostate Cancer Mortality and the Public Health Implication" https://www.nature.com/articles/s41598-020-63764-4
Hi,
I have a question about the script above. When I run it I get the error message:
I read a lot but I cannot find any solution. Has anybody an idea?
Thanks in advance.
Janita
This should have been a comment to the answer given above. You seem to have an encoding problem, does it really say
'Â'?yes it does. Do you have an idea how to fix this?
I have the same issue. Great post.
I can't guess what this might be. Other than making sure that I had gplots and devtools installed, I just copied and pasted the script below into R verbatim and it ran without error. At what point exactly does this error occur? The only thing I can guess is that it is some browser issue?
I run into the same problem too. I'm using Rstudio 0.98 and R 3.1.2.
I ran only the following code.
and got the error message
I searched a lot but can't find anything of
'Â'means. Has anyone solve this?Thanks,
Jack
I still can't really guess what is going on here. I just copied the code below into Rstudio 0.98 (R 3.1.2) and it ran without problems. If you type 'heatmap.3' I presume you see the function code has been successfully loaded into your session using source_url? Similarly, typing 'prob_matrix' produces sensible output? I'm guessing you have gplots and devtools installed and loaded or you would have gotten a different error. The only thing I can imagine is that your browser is doing something weird when you copy/paste the code from biostar and some hidden characters are coming along for the ride. Maybe try going directly to the source at github for both the function code and example script. See https://github.com/obigriffith/biostar-tutorials. You want Heatmaps/heatmap.3.R and Heatmaps/heatmap3_example.R. Safest might be to just clone the whole repo, navigate to the correct folder and then copy the function and script into R/Rstudio from a text editor you trust.
By copying the whole heatmap.3.R code fixed this problem. I think its because there is something wrong with the following code. But I'm not sure.
Thanks a lot for your help!