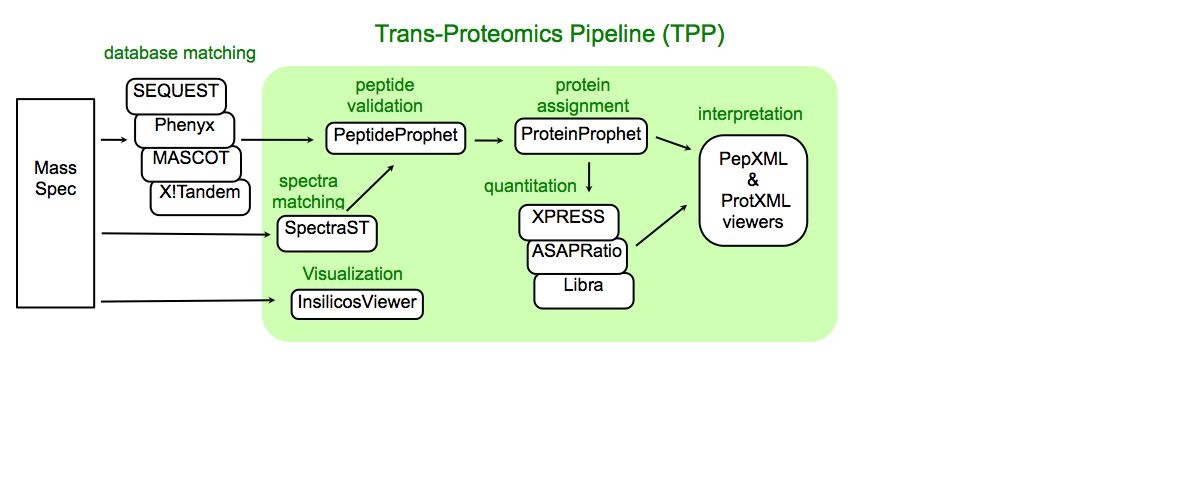
As we know the developement of high-throughput pipeline in different research field. Moreover, I am interested in knowing how high-throughput pipeline is deployed in Proteomics environment. One example of Pipeline I am going through is Trans Proteomics Pipeline (TPP)
I don't have any experince on workflow or pipeline design before; can you address
How can I manage reading, writing, schedular, execution and job failure operation in pipeline?
Hope to get some answers; though I am aware about the fact that Proteomics is discuss once in a year in Biostar!
I know this may not be a very useful answer to you, but I think the answer to your "how?" question is "with great pain and difficulty, if at all".
In my experience most of today's proteomics tools have not been built with pipelines in mind. Some run only on Windows platforms, some have no batch mode and have to be controlled through a GUI, and the output format of one tool will often not fit with the input format of what you want to run next.
Rather than attempting to build a fully automated pipeline from day one, I would first get an over view of which computational steps of the analysis flow currently takes the most manual work, and focus on getting those automated. You can then gradually automate more and more, and you may eventually reach the point of having a fully automated pipeline. You can be almost sure that the workflow itself will also be subject to change when new programs become available, so make sure to keep things highly modular to allow components to be swapped out without affecting the entire code base.
I think it varies very much between differnet "omics" fields. If you deal with microarray analysis, most tools exist in the form of R packages, and building a pipeline is thus essentially a matter of writing an R script that calls the right functions in the right order. If you work with sequence analysis, virtually all tools can be run from the command line and the file formats are very standardized; people thus typically write wrapper scripts around the tools and use a queueing system to distribute jobs on a cluster. Checking for job failure is inherently specific to each program.
Aside from the TPP, you might also want to check out OpenMS
In particular, OpenMS provides some quite good tools for MS1 processing (eg for label-free quantitation).
In my experience tools from OpenMS and from the TPP are quite reliable once you setup a workflow based on data from a particular instrument. The TPP are quite good at "failing" with appropriate error codes and messages when they encounter something unexpected.
A worse kind of failure (which you will find hard to check for in your pipeline) is that tools will run, but produce nonsense output. I think it's fair to say that some human checking is still required.
ADD COMMENT
• link
updated 5.9 years ago by
Ram
45k
•
written 14.4 years ago by
Ira
▴
20
0
Entering edit mode
Thanks but my quest was regarding reading, writing, executing and job failure issue!
I am in the process of open-sourcing our pipeline called Swift. http://goo.gl/aYJkJ
The idea was to make a tool that would be installable by mere mortals (I have so far to go still!) so you could deploy it yourself.
Sadly, this tool is very our-lab-specific. It requires Scaffold Batch 2 or 3 from Proteome Software, which many seem to be unable to afford.
I am working on making this tool more available by incorporating free peptide and protein prophet implementations from TPP. Alternatively, I could incorporate our work into TPP... not sure which way to go.
How was it done?
Pain, tears, code. Interfacing with third-party tools, especially badly documented ones, is major source of pain and frustration. The more complex your system gets, the more failures - and third party is not even under your control to fix issues you run into.
I would recommend you to work with somebody who did this before instead of rolling your own - it ended up being more work than I anticipated.




I think it varies very much between differnet "omics" fields. If you deal with microarray analysis, most tools exist in the form of R packages, and building a pipeline is thus essentially a matter of writing an R script that calls the right functions in the right order. If you work with sequence analysis, virtually all tools can be run from the command line and the file formats are very standardized; people thus typically write wrapper scripts around the tools and use a queueing system to distribute jobs on a cluster. Checking for job failure is inherently specific to each program.
That's why I dont wanted to be specific to Proteomics pipeline and asked question as a general in "omics" term.