
Hey guys,
Recently, I did some annotation of my de novo assembled transcriptome in a non-model plant RNA-seq project, using Sma3s.
I installed the softwares and modules required according to the documentation here.
Version:
Perl (v5.12.4),
BioPerl (1.006901, checked the version using the command perl -MBio::Root::Version -e 'print $Bio::Root::Version::VERSION,"\n"'),
CPAN (v1.9456),
local Blast (Blast 2.2.26, because Sma3s requires formatdb and blastclust, et al. scripts, so here is blast 2.2.26 rather than blast+),
the taxonomic division of UniProt database was plant division. I downloaded uniprot_sprot_plants.dat.gz and uniprot_trembl_plants.dat.gz, then uncompressed and used cat function to concatenate them to one file named uniprot_plants.dat for later use.
iMac, Mac OS X Lion 10.7.5, quad-core, 8 GB of memory.
The Sma3s.pl script can be found here at the bottom of the developer's site. The blast process went quite well, when it came to the annotation step, it appeared some error message like this below:
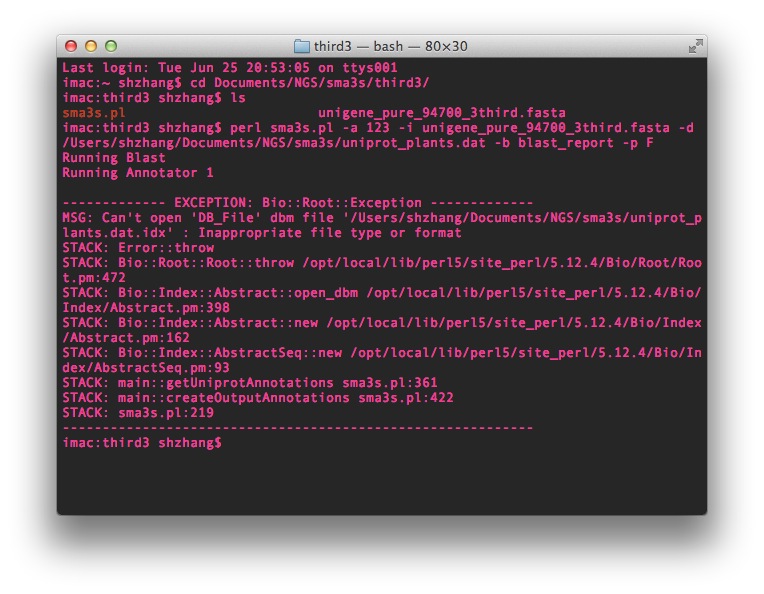
It first automatically generated some index files in the specific folder then ran blast process, these files contains:

I don't know what was wrong with my process, would you mind giving me a solution or tip?
Thank you.
Regards,
lzsph

Hi Evan,
It works well now.
Thanks!