
Forgive me if this is a dumb question but I assumed that the Average Heterozygosity was somehow related to the average distribution of frequencies seen for each allele in any given variation, i.e a AvHet close to 0.5 and a avHetSE lower than 0.1 would probably mean that that variation with 2 detected alleles would have a relatively balanced allele count for each like 0.45 for Allele A and 0.55 for Allele B.
Is my thinking flawed? I ask this because I filtered dbsnp137 using AvHet of >= 0.4 and avHetSE < 0.1 and I am getting loads of variations where 1 allele is clearly dominant with frequency count above 0.8.
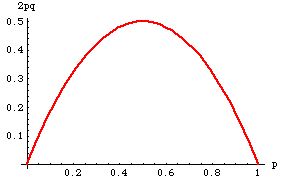
I've tried to get my head around the maths for the AvHet calculation in http://www.ncbi.nlm.nih.gov/projects/SNP/Hetfreq.html but I admit defeat. I am not a Mathematician by training and could not make sense of it.



Thank you for the detailed answers guys. They do make it much clearer but I am still perplexed why I am seeing such high allelic frequencies for the values of AvHet I had used to filter the dataset.
Take as an example this SNP:
ID: rs112111814 Alleles:C/T AlleleCounts:2137, 49 allele_frequencies:0.977585,0.022415 avHet:0.5 avHetSE:0
How can such a variation have a AvHet of 0.5 when 97% of seen alleles are C and less then 3% are allele T ?
It depends what you mean by 97% and 3%. Are these based on the 2137 counts? If yes, 3% amounts to 60 sequences. I don't know how many individuals you have nor your criteria for calling heterozygotes, but if you have 20 individuals, it is possible that in a few cases 50% of them will be called hetero (based on an average of 6 reads). In cases like this, I would suspect the presence of paralogs in your data set. For instance, you think that you are observing a single locus, but in fact the data from 2 different loci get combined. Locus 1 is 100% allele A and locus 2 is 100% allele B. This would give you high heterozygozities. In fact, when you do have paralogs, removing SNPs where Het is greater than 0.5 or 0.6 may help removing those paralogs.
See... I am now sure I am a complete ignorant because I cannot understand your explanation :(
The link to the variation is here: http://www.ensembl.org/Homo_sapiens/Variation/Explore?r=3:197694045-197695045;source=dbSNP;v=rs112111814;vdb=variation;vf=25173810
if contains 1092 individuals from 1K genome project with genotype calls: 1044 (C|C) / 48 (C|T)
The way I would look at this in the population tested there are 97% homozygotes (C|C) so according to your own graph the AvHet should be below 0.1 or am I just completely misunderstanding the calculations?
Thank you for your patience Eric
PS: this variation is a single locus.