I am using blastn to blast my data against tRNAs which is extracted from RFam database. Then I picked one match from the blast result to confirm it by directly searching my read in RFam online (http://rfam.sanger.ac.uk/search).
Here is my read which have a 100% match with a tRNA:
CCCATTCTTGCGACCCGGGTTCGATTCCCGGGCGGCGC
This is the matched tRNA from RFam:
GCGCCGCTGGTGTAGTGGTATCATGCAAGATTCCCATTCTTGCGACCCGGGTTCGATTCCCGGGCGGCGCA
Clearly it's a perfect match (in bold). But when you search my query in RFam, there is no hit. However, the subject tRNA can be found in RFam.
Could you paste the exact command you used for the BLAST? And the exact result line? Also, are you aware tha RFam provides a script for just this purpose? Here's the part of the code which is relevant:
Hi David, I will try the script you mentioned.
my blast command is : blastn -query mapped_reads.fa -task megablast -db tRNA_for_blast -out mapped_vs_tRNA.txt -outfmt "6 qseqid sseqid qlen evalue pident nident mismatch qcovs qcovhsp" .
The matched record is:
10002-681 2341-1 38 2e-15 100.00 38 0 100 100
The blantn result shows that it indeed is a perfect match. I am just wondering why this query read can not be found in RFam database. Is it too short?
I just skimmed through the script and the associated notes which I forgot to link to. Sorry!
It seem that blastn is just an initial step when querying the RFam database to narrow down the search. In the README, try jumping to the section (3) General comments on what rfam_scan.pl is doing. It seems that RFam does an additional step of matching the covariate models cmsearch (The run_multi_infernal_search() subroutine from the perl script.)
Thanks. It should be better using rfam_scan.pl . But I suppose that if a read is 100% match with a rRNA, then it should be from tRNA regardless of matching with CM model. I still want to know why my read can not be found by RFam online search.
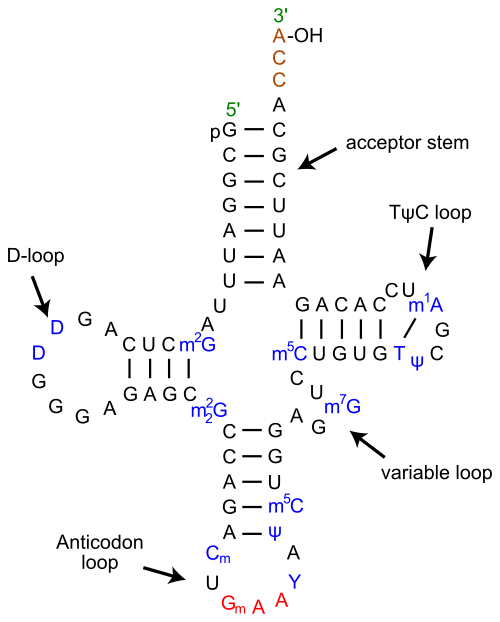
I think that it is because your tRNA sequence is truncated by having short reads. Covariance models use (among other things) information about the internal base-pairing of the RNA, in this case the cloverleaf structure.
(By Yikrazuul (Own work) CC-BY-SA-3.0, via Wikimedia Commons)
If you imagine an extreme case, where your tRNA fragment is exactly divided in half (this is almost the case for your example!), or you by chance get only a large part of the variable loop, then you
see that you loose a large portion of the most significant base pairing (acceptor stem, anticodon loop+ stem). That will possibly drop the score immediately,
because the structure left would be a single hairpin loop, which are quite frequent and not significant for a tRNA. That means that for truncated sequences, blast might be more sensitive than RFam or tRNAscan-SE.
You are right. My data is from miRNA sequencing, so the reads will all be short. In this way, as you suggested, blast should perform better than tRNA-scan or rfam_scan to find the tRNA sequences.


Could you paste the exact command you used for the BLAST? And the exact result line? Also, are you aware tha RFam provides a script for just this purpose? Here's the part of the code which is relevant:
$blastcmd = "blastall -p blastn -i $fafile -d $blastdb -e $blastcut -W7 -F F -b 1000000 -v 1000000 -m 8"; (By default $blastcut is 0.01)
Hi David, I will try the script you mentioned. my blast command is : blastn -query mapped_reads.fa -task megablast -db tRNA_for_blast -out mapped_vs_tRNA.txt -outfmt "6 qseqid sseqid qlen evalue pident nident mismatch qcovs qcovhsp" .
The matched record is: 10002-681 2341-1 38 2e-15 100.00 38 0 100 100
The blantn result shows that it indeed is a perfect match. I am just wondering why this query read can not be found in RFam database. Is it too short?
I just skimmed through the script and the associated notes which I forgot to link to. Sorry!
It seem that blastn is just an initial step when querying the RFam database to narrow down the search. In the README, try jumping to the section (3) General comments on what rfam_scan.pl is doing. It seems that RFam does an additional step of matching the covariate models cmsearch (The run_multi_infernal_search() subroutine from the perl script.)
Thanks. It should be better using rfam_scan.pl . But I suppose that if a read is 100% match with a rRNA, then it should be from tRNA regardless of matching with CM model. I still want to know why my read can not be found by RFam online search.
Have you tried contacting the lovely Rfam folks with this question? (http://rfam.sanger.ac.uk/help#tabview=tab12)
Thanks for your advice. i really should try to contact them.