
I am frequently faced with variant files of either 0-based or 1-based coordinates (or in the worst case, mixed!) and having to determine which I am looking at and how to convert between them. I usually go to the white board and work it out. This time, I figured I would just create a digital copy.
In addition to the illustrations/explanation below, Ben Ainscough (a member of our lab) has created a python tool to convert between zero and one based coordinate systems here: https://github.com/griffithlab/convert_zero_one_based
First, a diagram to help illustrate:
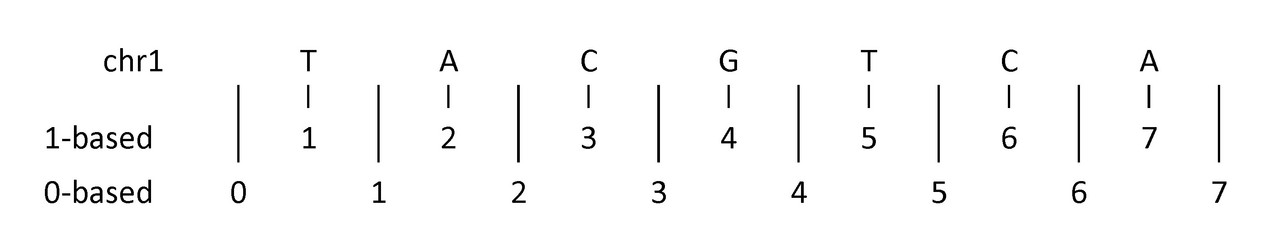
The example above shows (an imaginary) first seven nucleotides of sequence on chromosome 1:
- 1-based coordinate system
- Numbers nucleotides directly
- 0-based coordinate system
- Numbers between nucleotides
To indicate a single nucleotide or variant:
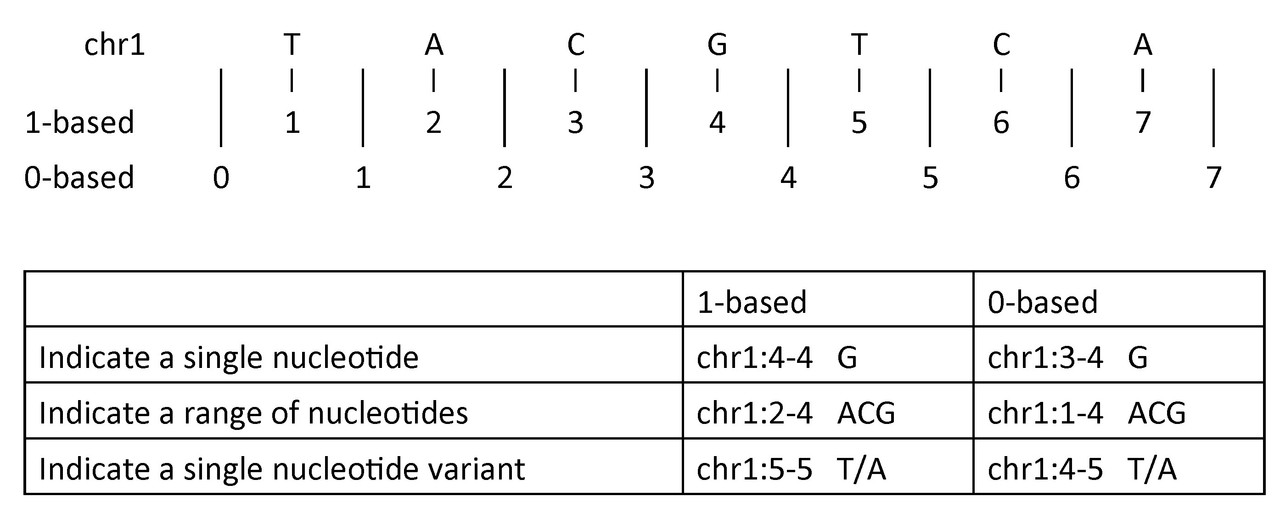
- 1-based coordinate system
- Single nucleotides, variant positions, or ranges are specified directly by their corresponding nucleotide numbers
- 0-based coordinate system
- Single nucleotides, variant positions, or ranges are specified by the coordinates that flank them
To indicate a deletion or insertion:
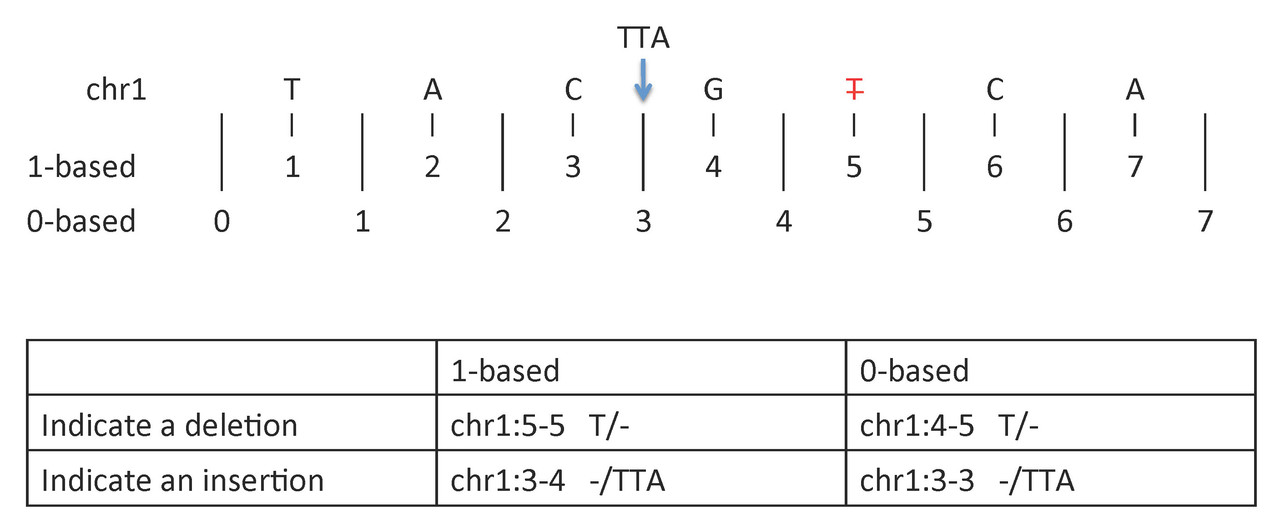
- 1-based coordinate system
- Deletions are specified directly by the positions of the deleted bases
- Insertions are indicated by the coordinates of the bases that flank the insertion
- 0-based coordinate system
- Deletions are specified by the coordinates that flank the deleted bases
- Insertions are indicated directly by the coordinate position where the insertion occurs
Why does all this matter?
- Moving from UCSC browser/tools to Ensembl browser/tools or back
- Ensembl uses 1-based coordinate system
- UCSC uses 0-based coordinate system
- Some file formats are 1-based (GFF, SAM, VCF) and others are 0-based (BED, BAM)
- See this excellent post and its many good links for more info:
Pseudo-code to convert variants from 0-based coordinates to 1-based coordinates:
if (type=SNV){start=start+1; end=end;}
if (type=DEL){start=start+1; end=end;}
if (type=INS){start=start; end=end+1;}
Pseudo-code to convert variants from 1-based coordinates to 0-based coordinates:
if (type=SNV){start=start-1; end=end;}
if (type=DEL){start=start-1; end=end;}
if (type=INS){start=start; end=end-1;}

GBrowse and GFF are 1-based exclusively.
I'm not sure your description of an insertion in 1-based coordinates is canonical. I'm actually not sure there is a canonical description of insertions in 1-based coordinate range, which is why many low-level tools prefer to work in 0-based coordinates.
One of the primary advantages of 0-based coordinates is that the width of a feature is always 'end-start'. Insertions have 0-width, and hence they have the same start/end.
Your representation in 1-based coordinates makes the insertion appear as if it has width of 2! I'm not sure there is a better representation, but more programmatically friendly is to describe it as chr1:5-4 (yes, start > end). It gives you the advantage of the width of the feature always being (end-start+1) and translation from 0 to 1 base only require incrementing the 'start' for ALL variant types.
Because of these problems, I would argue for variants, you should not represent them in 1-based coordinates as a range, but just as the start coordinate only.
In ENSEMBL they treat the INS giving start>end: http://www.ensembl.org/info/docs/tools/vep/vep_formats.html#vcf
The following examples illustrate how VCF describes a variant and how it is handled internally by VEP. Consider the following aligned sequences (for the purposes of discussion on chromosome 20):
Individual 3
The third individual has an "A" inserted between the 3rd and 4th bases of the sequence relative to the reference. In VCF, as for the deletion, the base before the insertion is included in both the reference and variant allele columns, and the reported position is that of the preceding base:
In Ensembl format, again the preceding base is not included, and the start/end positions are "swapped" to indicate that this is an insertion. Similarly to a deletion, a "-" is used to indicate no sequence in the reference:
Again, the output will appear different, and the constructed identifier may not be what is expected:
The solution is to always add a unique identifer for each of your variants to the VCF file, or use VCF as your output format.
The gff3 spec indicates insertion sites should have start = end and the insertion occurs to the right of the coordinate. It's a little goofy but at least it's a "standard".
I agree with regard to zero based coordinates: both chado and jbrowse use zero based internally.
Yes. Most of what you say is absolutely true. Except I'm not sure about your last point. How would you indicate a multi-bp deletion in 1-based with only a start coordinate? In any case, the point of this cheat sheet was not to propose a standard but merely to describe the real world problem. As someone working at a genome center and dealing with a large variety of standard and non-standard variant files it is quite common to see both 1-based and 0-based variant files with chr:start-stop format whether that makes sense or not. The main thing is to realize that you need to think about this issue before blindly passing a variant file to any piece of software.
Completely agree that there is under-appreciated complexity in different and (sometimes conflicting) representations used by different tools.
My last point is simply this: 1-based coordinates are inadequate to describe variants as "features" as a pair of start/stop 1-based indexes (because of the issue of insertions having 0-width). Yet of course, we sometimes are faced with the need to attempt to do so, and I actually prefer the (start+1,end) representation precisely as it gives the impression that you are representing something unnatural in 1-based coordinates when you see chr1:5-4.
It also is capable of being round-trip converted back to 0-based coordinates without knowledge of what type of variant the coordinates represent as it is variant type agnostic.
But for human readable consumption, 1-based is preferred and my preference is to write variants then with just the start 1-based and use something akin to (or equivalent of) HGVS g. notation.
For example:
When representing variants in a format intended for programs to parse though, 0-based intervals are always preferred.
You write that UCSC is 0-based. As has been mentioned above, the web-interface of the UCSC browser is 1-based, as it meant to be used by biologists, but all internal representations (text files, database tables, binary files) are 0-based, as they're mostly used by programmers.
Unfortunately, for historical reasons, there two exceptions, two formats (the wiggle and bigWig text file and database formats) are 1-based.
see here https://genome.ucsc.edu/FAQ/FAQtracks#tracks1 Database/browser start coordinates differ by 1 base
scholar alert: this post was cited in :
Fun fact regarding IGV: files with .igv ending are 0-based, while the IGV genome browser itself uses the 1-base to display sequences 🤦🏻♂️
(I see @Maximilian Haeussler mentioned the same regarding UCSC)
It doesn't make sense to create such two coordinate systems. One unified system would make it easy to use and do not make us to make mistake.
That ship has long ago sailed (heck, even fortran and C differ in whether to use 0 or 1 based indexing by default).
Yes, you are right. perl and python use 0 as index of the array.Anyway, our programmer need to remember all the traps. Maybe it is a natural barrier to stop the non-programmer enemy come to our field easily. LoL. Anyway. I like it.
Actually, in genomics, it has a clear advantage of making interval calculations straightforward (end - start).
While BAM is 0-based, once you pull it out to something human-readable it can get turned into 1-based data (SAM). Great post, btw.
Is it true that SAM is 1-based but BAM is 0-based? Why would it be designed this way?
BAM Is zero based. : https://samtools.github.io/hts-specs/SAMv1.pdf
I would say because C programmers like starting things with zero. Anyway, who cares ? unless you're writing your private BAM parser people use an API to read the BAM data. For example
htsjdk/SAMRecord.getAlignmentStart()returns the 1-based POSThanks. My confusion mainly came from if BAM is 0-based, then it would make more sense if SAM is also 0-based since the two are essentially the same except for one is compressed while the other is not. But as you explained, if that's not case, it's very unintuitive to me, but very good to know.
This is super useful, thanks. I've found another nice tutorial covering this topic nicely: http://genome.ucsc.edu/blog/the-ucsc-genome-browser-coordinate-counting-systems/
and some API are 1-Based ! e.g: the java picard library for BAM use 1-based indexes :-)
For god sake, check the TAIR10 Gbrowser, it just like 0-based/1-based mixture to me (see post Help: TAIR10 GBrowser 1-based mixed with 0-based?). Luckily we got IGV. (p.s. Plant research cannot triumph mammalian research, look at ucsc genome browser.)
I came back to this which I had bookmarked but all the images have disappeared. Any chance of bringing them back?
Images are still there. Browser issues perhaps?
The images are blurred...
Images are gone for me, too, FWIW. Resorting to Wayback Machine.
Still there for me. This sounds like a BioStars or system-specific issue?
No, the images are gone for me as well. Perhaps you have them cached locally.
If you have time to do a solid for everyone who visits this question, maybe grab a copy of those images, upload them to another server, and edit the question to point to the new image host?
Hm, they're here for me now, again. Either (1) he moved them to a better host, or (2) the host is just unreliable and goes down at various times / has issues with its CDN that mean it works for some locations but not others. Leaning towards (2), as as far as I can tell from the Wayback post, the host is the same (http://postimages.org/).
postimages.org has been having some issues with their hosting service (and business model for that matter). The domain name postimg.org was apparently locked by registry. They moved to postimg.cc. Once I updated the urls they returned. BioStars developers have been informed about this as I suspect it is a site-wide issue since this is a popular choice for free image hosting.
Something wrong about Pseudo-code of converting INS between 0-based and 1-based. In 0-based system, INS coordinate is usually in the format of [X,X), where X is a coordinate. In 0-based, [X,X) INS means sequence is inserted before X, while X is not include. In 1-based system, there is no such mechanism, so you have to convert to [X+1,X+1] to indicate that the sequence is inserted before X+1. If you convert to [X,X+1] as you described above, it makes no sense.
The same thing happened when converting from 1 to 0-based.
So to convert INS from 0 to 1-based system:
start = start + 1; end = end + 1
and to convert INS from 1 to 0-based system:
start = start - 1; end = end - 1
For a further discussion, INS is a very tricky case. Some program does not recognize coordinate like [X,X) in 0-based. So there is another representation of [X,X+1) which also indicate that sequence is inserted before coordinate X. In this situation, the converting will be as follows:
to convert INS from 0 to 1-based system:
start = start + 1; end = end
to convert INS from 1 to 0-based system:
start = start - 1; end = end
For me I vote the second representation, which will unify the converting method.
But I still did not come up with the case you offered above about INS converting.
Just thought I'd add that the UCSC browser is 1-based, while its tools are 0-based.
Incidentally, this makes no mention of zero-based, half-open coordinates, which are convenient. Complete Genomics used to use them. My first variant-processing programs used zero-based, closed coordinates, but later I found that half-open is easier to deal with. So, BBMap's current variant format uses zero-based, half-open as it seems to be the most efficient. 1-based formats always cause problems, so I consider it unfortunate that they were selected for sam and vcf.