Entering edit mode

11.0 years ago
Rnaer
▴
120
From all the possible seqeunces of 30 nt (4 ** 30 possibilities), how can I quickly find the ones that does not match to any region of the C.elegans genome. Is there any tool to do that?


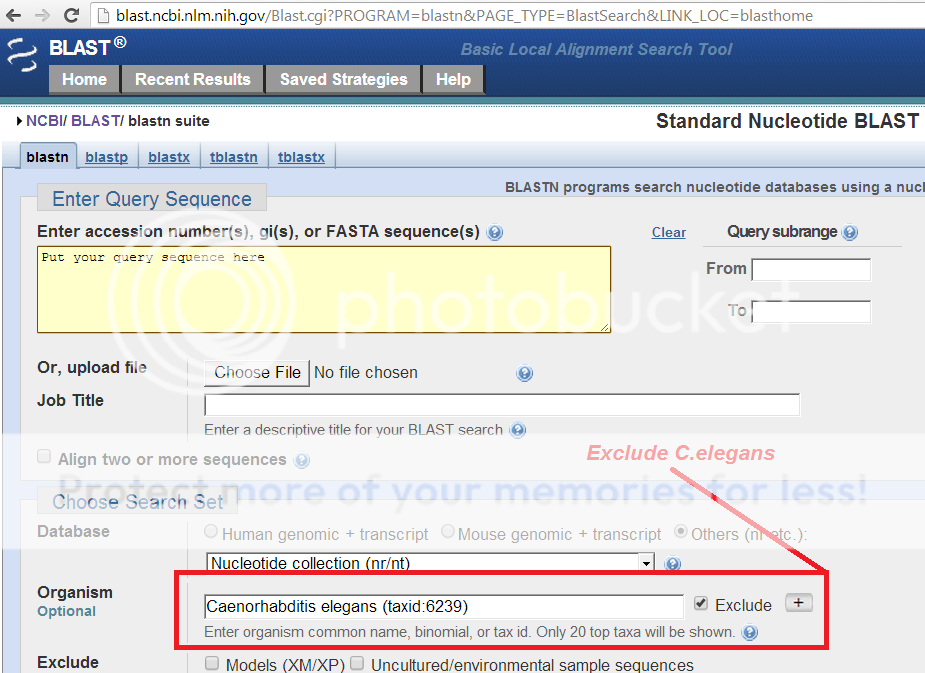

Does it mean you want to compare 4^30 sequences against C.elegans genome ?
yes, basically, but of course, not do it in brute force way
No...I don't think you really want to check every single 30-mer in existence. It would take a thousand years. Why don't you start by generating, say, 1000 random 30-mers, and aligning them to your genome, and seeing if the worst aligning ones don't suit your needs.
What about my answer, is this what you want?
I am afraid that your answer is not what he is looking for.
How many of those do you want? (All?)
well, ideally, the most distant sequences from any regions of the target sequence, but one is enough
If I understand you correctly, than I would probably just generated 100k random sequences of length 30 and for every 30nt window of C.elegans performed Needlman-Wunsch algorithm with all my 100k sequences and reported the worst alignment.
yes, but that's too brute force. I actually agree with swbarnes2. It's not so trivial to implement it, though. just want to see if any tool exist.
Why don't you use bowtie? It's quite fast and you can use it with fasta sequences of the 30-mers. You can set the parameters to be exact matches and allow for multi-mapping. Then just look for the 30-mer sequences (reads) that don't align to the genome.
even each 30mers takes 1 sec, to test all possibilities will take 4**30/3600 = 3.20256e+14 hrs
You know that short read aligners work much faster than 1 read every second. Bowtie aligns tens of millions of reads in hours. So aligning 100K test reads would be feasible.
if you wanna check for k-mers and which of them occur just go with Jellyfish