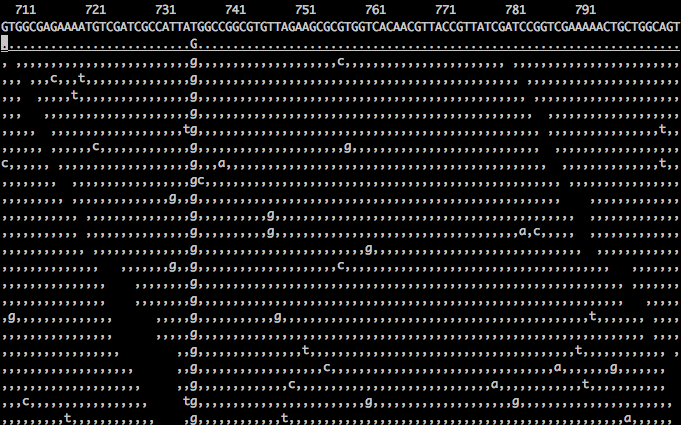
As you can see in the screenshot below, there are many mismatches (sequencing errors) but there is only one SNP. (image via http://biobits.org/samtools_primer.html)
It gets especially fun when you are sequencing a population, and some of those singletons represent true variation within the population, very difficult to separate sequencing errors and population variation.
Because the veracity of base calls in NGS isn't 100% (not to mention that alignment isn't always perfect). So, maybe the mismatch is a technical artifact, or a PCR error, or something else entirely. That's why you need multiple non-PCR duplicate sequences covering a position to confidently call a variant there. I should note that this was also the case in the pre-NGS days of Sanger sequencing, so it's not particular to NGS.
A mismatch is a difference of one (or more) bases of a sequences compared to the reference genome, in an alignment.
A SNP is a position in the genome that is known to vary among individuals of the same species, or of the same population.
In the pre-sequencing era, defining SNPs was a expensive and long process in which candidate SNPs were first sequenced in a sample population, as a way to select only the positions that effectively varied with a certain frequency, and that were representative of variation in the population. This process eliminated all the variants that were observed with a low frequency (e.g., a mismatch in one individual out of hundreds was not considered enough), and also discarded all the tri-allelic variations (note that all SNPs used to date are bi-allelic by definition).
In NGS time, I think that most SNP calling algorithms require more than one single mismatch in a position to define the position as a SNP. Thus, in order to be considered a SNP, a position must show the same mismatch in more than a few samples. Moreover, positions that show more than two loci are also discarded as SNPs, as we are only interested in bi-allelic SNPs.
a sequence mismatch is a NECESSARY BUT NOT SUFFICIENT condition for a SNV
put in other words, a SNV requires a mismatch, but not all mismatches are SNVs
side note: when sequencing a single sample one can't really say that it detects a SNP, but a SNV instead (Variation instead of Polymorphism). that variation must be checked against population frequencies databases in order to confirm whether that variation has been uniquely detected on that sample, or whether that same variation has been reported previously on other samples. based on this reported frequency, if - strictly speaking - it's above 1% then the term SNP can be appropriately used.





It gets especially fun when you are sequencing a population, and some of those singletons represent true variation within the population, very difficult to separate sequencing errors and population variation.