I have just started to analyze an RNAseq dataset. When I run FastQC on my fastq files, I see a problem with "Adapter Content" I have attached the image of the Adapter content.
I expected to see the adapter sequences in the first positions of each read [or I am wrong??], but they show up in the end portions of the reads. I tried Trimmomatic for clipping the adapters (ILLUMINACLIP:TruSeq3-PE.fa:2:30:10), but it didn't solve the problem, and I see a similar figure after Trimming. What is wrong with my approach? What should I do?
Nothing, the adapter sequence file provided by Trimmomatic must just not include your adapter sequence. Read their documentation to determine if you're using the right file for your adapters and adjust if necessary. Or use TrimGalore or such that include more general adapter databases.
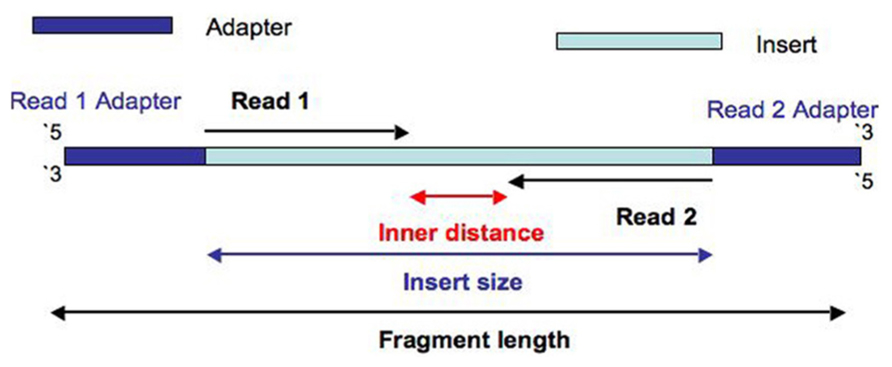
You will see adapter content at the end of reads usually. If you have them at the beginning, that's indicative of adapter dimers, which are a very not good thing and usually handled easily during size selection and library clean-up. Adapter content is found when the read length is greater than the insert size, resulting in the read running into the adapter sequence. I found this image helpful to visualize how this occurs:
Adapter content is easily handled, but it's wasted sequencing. This can be mitigated by altering the fragmentation protocol to slightly increase fragment size, as the standard protocols tend to result in a decent amount of fragments smaller than the read length if it's 150 bp. The library prep protocols from Illumina, etc, will have an appendix that explains how the fragmentation can be adjusted to alter fragment size distributions.
I expected to see the adapter sequences in the first positions of each
read [or I am wrong??],
In case of Illumina sequencing you will always see adapters at the end of the reads. This happens in cases where you run out of insert to sequence (short inserts) and start sequencing into adapter on 3'-end. Illumina sequencing primer is located such that you start sequencing your insert on 5-end starting at first base.
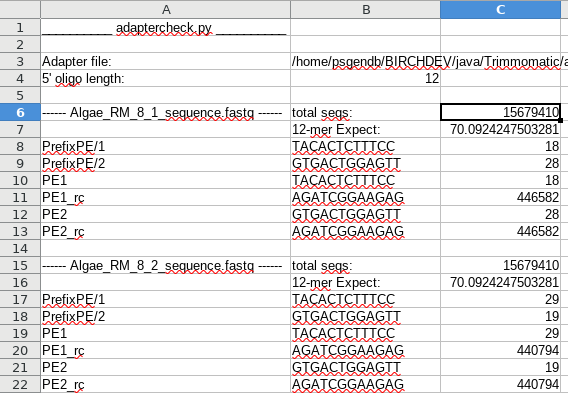
The latest release of BIRCH, v3.80, has a new program called adaptercheck.py which generates a spreadsheet that lists, for each fastq file, the adapters supplied from a fasta file, the number of hits of size k expected, and the number of hits actually found:
I wrote this program when faced with a set of fastq files for which there was no record of which adapters were used. It turned out to be the best way to find out which adapters had been used, and also to assess the amount of read-through.
By the way, if these are recent RNAseq reads, the apparent insert size is a bit disappointing. With the ancient Illumina reads I had to work with, I only saw a few percent of reads with 3' adapters. If your RNAseq reads are presumed to be 100 nt, my guess is that the library size falls well short of that.
You may also be interested to know that BIRCH lets you run just about anything you need for processing reads (FastQC, Trimmomatic, trim_galore ... ) using our point and click BioLegato family of applications.





Thanks a lot for the clear explanation.