Entering edit mode

3.4 years ago
pavelasquezv
▴
50
Hi all,
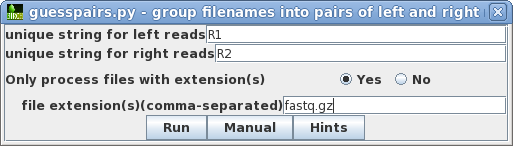
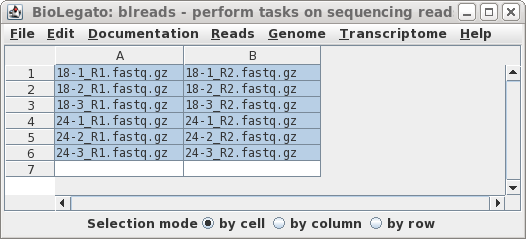
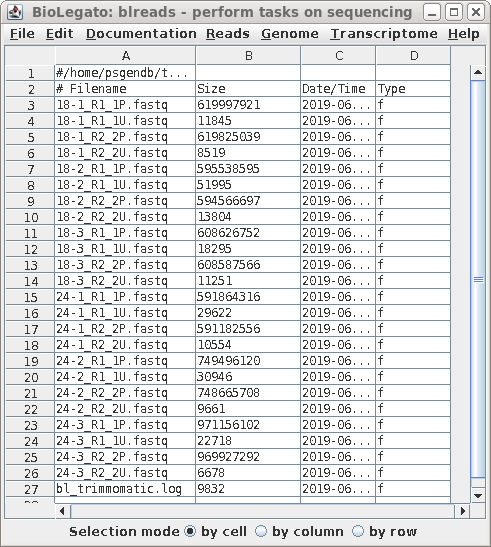
I have to run the same program for multiple files.
I would like to know if I can do that using a loop?
Many thanks, friends!
Files: SRR10345445_1.fastq SRR10345445_2.fastq SRR10345446_1.fastq SRR10345446_2.fastq
TrimmomaticPE -threads 30 \
SRR10345445_1.fastq SRR10345445_2.fastq \
SRR10345445_1_PE.fastq SRR10345445_1_SR.fastq SRR10345445_2_PE.fastq SRR10345445_2_SR.fastq \
HEADCROP:12 ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10:2:keepBothReads \
SLIDINGWINDOW:4:20 LEADING:5 TRAILING:5 MINLEN:40
TrimmomaticPE -threads 30 \
SRR10345446_1.fastq SRR10345446_2.fastq \
SRR10345446_1_PE.fastq SRR10345446_1_SR.fastq SRR10345446_2_PE.fastq SRR10345446_2_SR.fastq \
HEADCROP:12 ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10:2:keepBothReads \
SLIDINGWINDOW:4:20 LEADING:5 TRAILING:5 MINLEN:40