
Hi
I am doing a microbiome ordination analysis and I am not entirely sure on the use of agglomeration of taxa for this ordination analysis. I have a dataset with amplicon sequence variants that are assigned up until genus level. I wondered whether it makes sense to use agglomerated taxa for this ordination analysis and, as well in further steps (normalization, diversity analysis)? Why (not)?
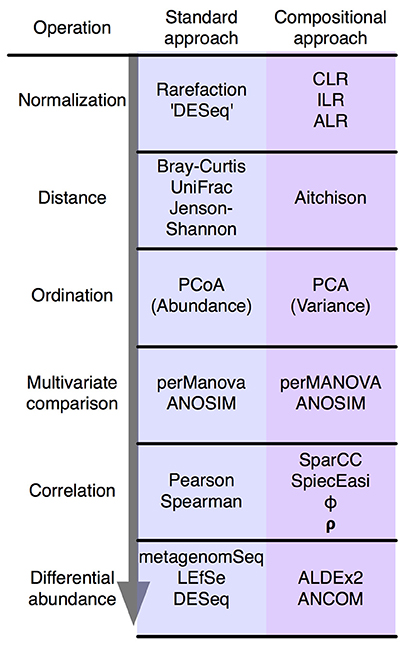
Also, more on this ordination analysis, I am following this figure from https://www.frontiersin.org/files/Articles/294209/fmicb-08-02224-HTML/image_m/fmicb-08-02224-g002.jpg of which I derive that, when using rarefied counts (which I do use in my analysis), I need to follow the approach and use the Bray-Curtis dissimilarity distances as well as PcoA. However, when doing so, I got an error:
ps_of_choice <- ps_rare
method_of_choice <- "bray"
ord_of_choice <- "PcoA"
dist_matrix <- phyloseq::distance(ps_of_choice, method = method_of_choice)
dist_matrix <- as.matrix(dist_matrix)
head(dist_matrix)[,1:6]
ord <- phyloseq::ordinate(ps_of_choice, ord_of_choice)
phyloseq::plot_scree(ord) +
geom_bar(stat="identity", fill = "blue") +
labs(x = "\nAxis", y = "Proportion of Variance\n")
head(ord$CA$eig)
sapply(ord$CA$eig[1:5], function(x) x / sum(ord$CA$eig))
#Scale axes and plot ordination
clr1 <- ord$CA$eig[1] / sum(ord$CA$eig)
clr2 <- ord$CA$eig[2] / sum(ord$CA$eig)
phyloseq::plot_ordination(ps_of_choice, ord, type="samples", color="gender") +
geom_point(size = 2) +
coord_fixed(clr2 / clr1) +
stat_ellipse(aes(group = gender), linetype = 2)
Error in unit(abs(aspect_ratio), "null") : 'x' and 'units' must have length > 0
If I change the ord_of_choice (see line ord <- ...) to RDA, then the error is no longer there. Why is this the case? I have the code from an online tutorial, and I think the error must be in these "clr1" and "clr2" lines, but I don't understand what is exactly happening there.
Thank you in advance!

{kind=link}
It looks like your question changed while I was writing a response. I will leave it here in case you find it helpful.
Yes, I have figured out already a bit on this agglomeration approach, but I'm still a bit stuck on this ordination analysis. I will edit my question to also this agglomeration question so that your answer replies on this part as well.