Entering edit mode

5.9 years ago
s.m.frampton
▴
20
Hello,
I am very new to the world of sequencing and would really appreciate your knowledge. I am trying to study a genomic region containing 5 very homologous genes and have obtained the FastQ files generated using MinION. I used the tool NanoPlot to produce a QC report but am struggling in understanding it. Particularly regarding the quality scores and quality cut-offs; I appreciate a quality threshold depends upon my application but would it be possible to explain very simply what the number of the 'mean read quality' actually represents? If I go on to filter my reads, is there a standard cut-off for ONT data?
Any information around this would be extremely helpful!

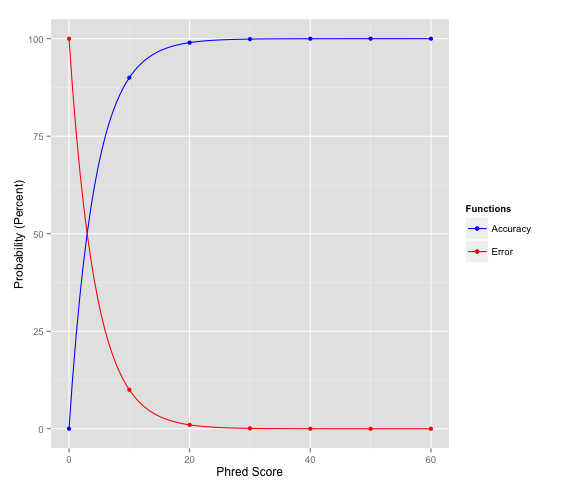
Wikipedia is also not bad here, but it might be tricky to find the right page.
https://en.wikipedia.org/wiki/FASTQ_format
A rough estimated might by PHRED quality scores of ~8-12 for raw nanopore reads and ~30-35 for illumina reads.
Hello!
I have obtained a 'Median read quality' value of 9.4 for a metagenomic sample through Nanoplot analysis. Is there any mathematical formula for converting this value to a Phred score?
Regards
9.4 is the phred score
Thanks a lot, sir!
Here is a snapshot of another part of my NanoPlot result:
Number, percentage and megabases of reads above quality cutoffs
Please suggest:
Thanks and Regards