Hi Kermit and prash (and others)!
Nice discussion. I wanted to offer some biological rationales in addition to the technical reasons that Prash has already mentioned.
Background:
First, note a VCF file is large in proportion to the number of differences it has relative to the reference genome(s) being used, because only a difference to the reference is recorded in a VCF and thus gets a new line, lengthening the file.
In a constitutional (germline) setting:
Since most are still using GRCh38, the reference is primarily a person of European ancestry. As such, relative to such a reference, persons of African or Asian heritage may have more genetic variants. This also applies to persons having genetic admixture, who tend to have more sites of heterozygosity on average than persons whose ancestry reflects one single population.
In a somatic setting:
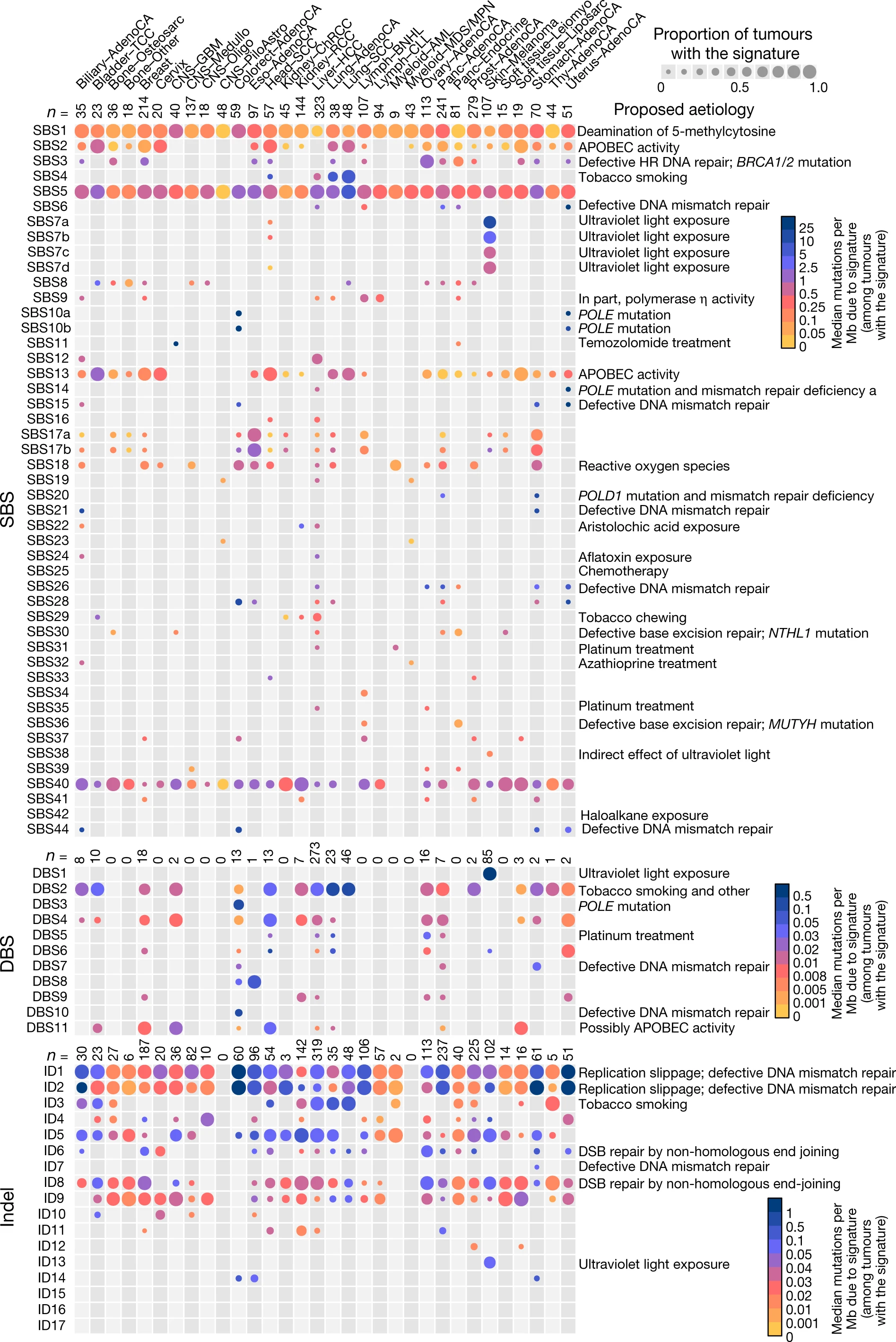
Now, you mentioned TCGA, which is a cancer database. Depending on the driver mutations, the somatic mutation rate varies tremendously both within and across cancers. Figure 3 of alexandrov et al., 2020, illustrates this very well. In case that picture is inaccessible, the PMID is 32025018. When I say tremendously, it is not an exaggeration; on one extreme you have for instance Polymerase Q driven hypermutatory phenotype, the other extreme might be certain kinds of Oligodendroglioma, whose somatic mutation rate is most often low, rather, it is thought that epigenetic and other changes do much of the work in "turning the cells malignant" so to speak. The VCF file will, of course, grow in size in proportion to the number of genetic variants that present. Therefore, the VCF for a PolQ driven malignancies may be larger than many of the oligodendroglioma tumors, and also larger than a paired normal VCF file.
Returning to the prior answer; technical reasons:
More specifically, the VCF file will grow in proportion to the number of variants that can be called accurately, which typically means having an appreciable allele frequency. This returns us to Prash's ideas, relating to the many technical reasons why they differ in size. As already indicated, the higher the read depth of the genome (i.e., 10x, 100x, 1000x, 10000x) sequencing, the lower the variant allele fraction that can be called at a given level of confidence. This is important in a somatic setting, because intratumoral heterogeneity is in some cases predictive of prognosis, treatment failure, etc. Therefore, it can be important to capture the variants found in a subclone of a tumor even if, presently, it does not account for a large percentage of the cells in the tumor.
Other important technical factors include, but are certainly not limited to: sample quality (which might vary depending on formalin fixation, age, etc), sample processing pipeline, and the tumor percentage of the sample (which directly relates to the VAF detection problem, above).
Big picture:
Of these factors, the somatic changes will tend to affect the size of the VCF more than the differences in germline variation relating to ancestry or what have you, because the somatic mutation rate can get so high. Also, of note, these factors apply not only to WES, but WGS or other designs as well.



{kind=link}
"It's not a gvcf with site coverage for the entire cohort, I called the variants myself" This sentence doesn't make sense to me. gVCFs are generally single-sample. are your vcf(s) single-sample?