Entering edit mode

11 weeks ago
Vijith
▴
90
I have two queries: one is regarding AUGUSTUS, and the second is about extracting sequences from the *.gff file for downstream BLASTx homology-based annotation.
- I ran AUGUSTUS using the command
augustus [parameters] --species=SPECIES queryfilename > output.gff
without specifically setting --alternatives-from-sampling to True. Will this affect the completeness of the downstream annotation process?
- After completing the de novo gene prediction, I want to run BLASTx for homology-based annotation, alongside the evidence-based approach. However, I’m confused about extracting features from the *.gff output. In a previous faulty AUGUSTUS run, I used AGAT's
**agat_sp_extract_sequences.pl**to extract sequences with the command.
agat_sp_extract_sequences.pl -g infile.gff -f infile.fasta -t gene
However, after reading more about its suitability for BLASTx, I realized this approach might include introns, UTRs, intergenic regions, etc. Therefore, I’m considering using the alternative command:
agat_sp_extract_sequences.pl -g infile.gff -f infile.fasta --mrna
or
agat_sp_extract_sequences.pl -g infile.gff -f infile.fasta -t cds.
If you want details on what these commands extract, please take a look at this image.

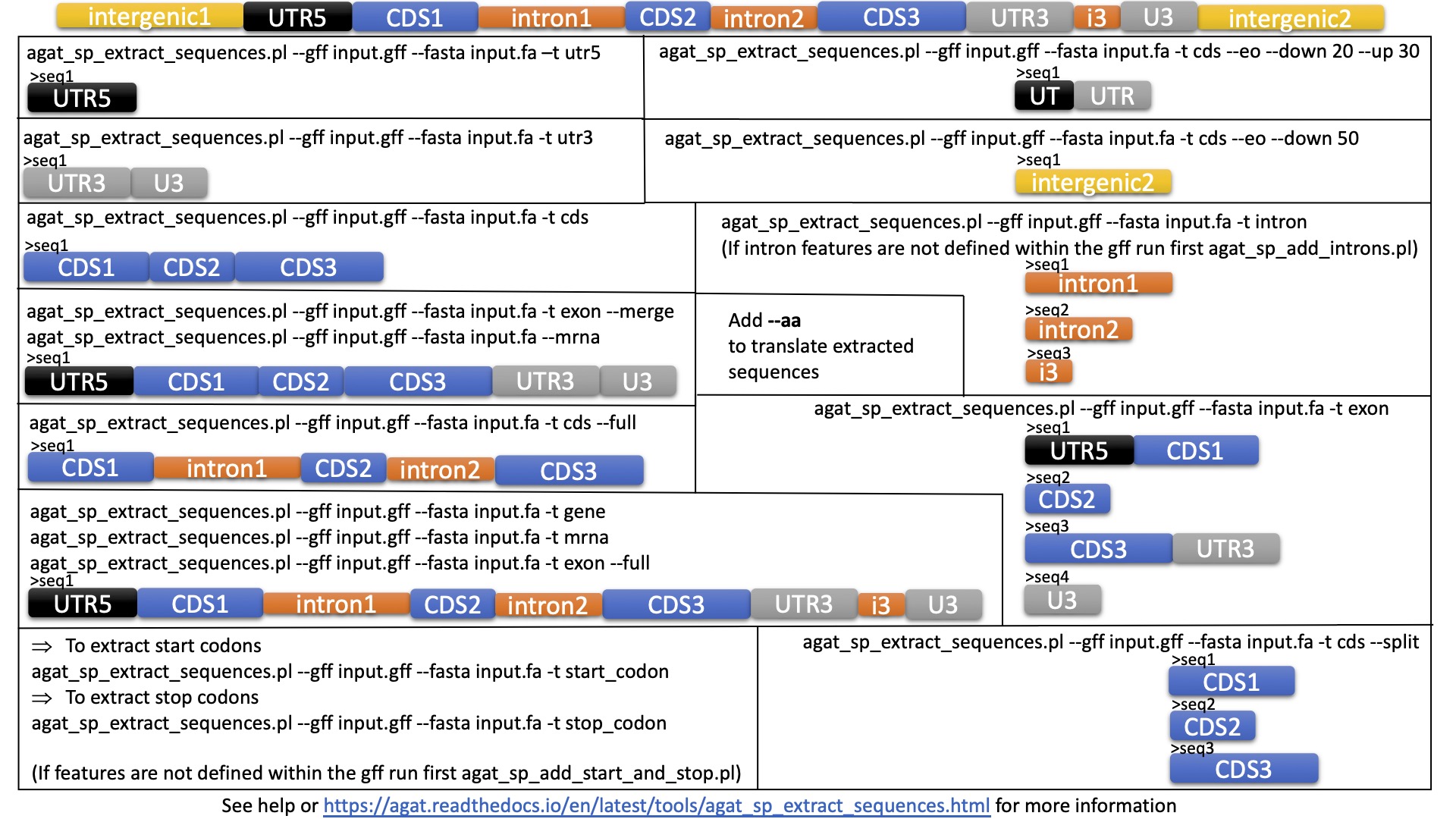{kind=link}
Juke34, thank you so much for your valuable response. This helps.