Inspired by the limitations of the approaches I mentioned above, I just released a new tool called multiIntersectBed in bedtools version 2.14.3. I realize that this solution doesn't address your request for 50% reciprocal overlap, but I can't yet envision an efficient way to do that other than what has already been proposed.
The basic concept of this approach is that it compares the intervals found in N sorted (-k1,1 -k2,2n for BED) BED/GFF/VCF files and reports whether 0 to N of those files are present at each interval.
An example is likely best to illustrate what the tool does. First, here's a graphical representation:
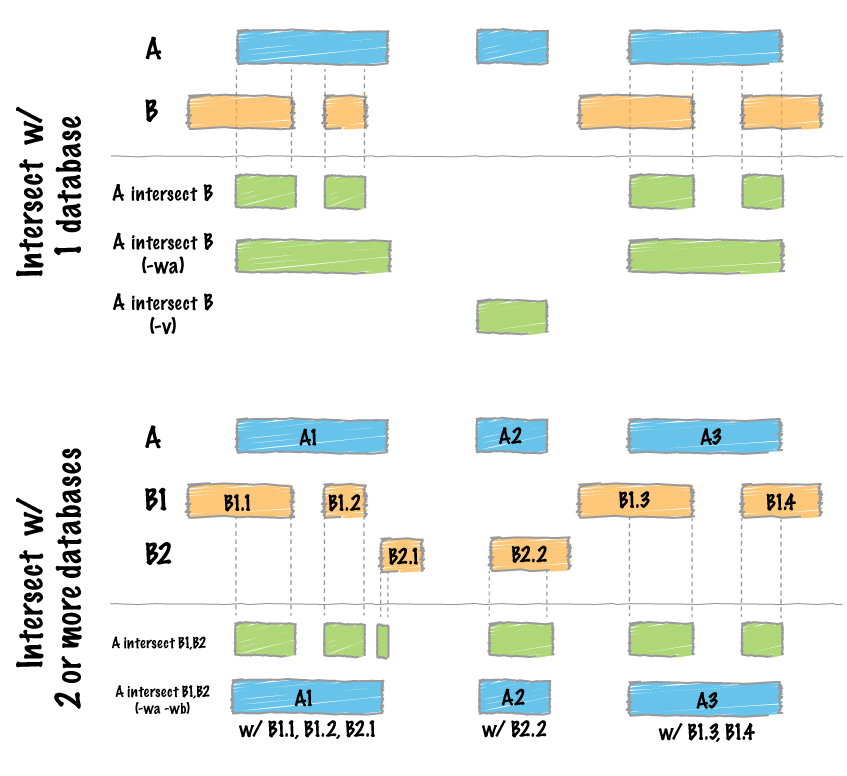
Now, an example with real BED files and real output.
$ cat a.bed
chr1 6 12
chr1 10 20
chr1 22 27
chr1 24 30
$ cat b.bed
chr1 12 32
chr1 14 30
$ cat c.bed
chr1 8 15
chr1 10 14
chr1 32 34
In the example below, the first three columns define the interval, the fourth column reports the number of files present at that interval, the fifth column reports a comma-separated list of files present at that interval, and the 6th through 8th columns report whether (1) or not (0) each file is present. The order is the same as on the command line.
$ multiIntersectBed -i a.bed b.bed c.bed
chr1 6 8 1 1 1 0 0
chr1 8 12 2 1,3 1 0 1
chr1 12 15 3 1,2,3 1 1 1
chr1 15 20 2 1,2 1 1 0
chr1 20 22 1 2 0 1 0
chr1 22 30 2 1,2 1 1 0
chr1 30 32 1 2 0 1 0
chr1 32 34 1 3 0 0 1
The above example only reports intervals where >=1 file has coverage. We can also get a complete picture of the chrom by using the -empty parameter and by providing a genome (chrom sizes) file:
$ multiIntersectBed -i a.bed b.bed c.bed -empty -g genomes/human.hg18.genome
chr1 0 6 0 none 0 0 0
chr1 6 8 1 1 1 0 0
chr1 8 12 2 1,3 1 0 1
chr1 12 15 3 1,2,3 1 1 1
chr1 15 20 2 1,2 1 1 0
chr1 20 22 1 2 0 1 0
chr1 22 30 2 1,2 1 1 0
chr1 30 32 1 2 0 1 0
chr1 32 34 1 3 0 0 1
chr1 34 247249719 0 none 0 0 0
We can also get a header:
$ multiIntersectBed -i a.bed b.bed c.bed -empty -g genomes/human.hg18.genome -header
chrom start end num list
chr1 0 6 0 none 0 0 0
chr1 6 8 1 1 1 0 0
chr1 8 12 2 1,3 1 0 1
chr1 12 15 3 1,2,3 1 1 1
chr1 15 20 2 1,2 1 1 0
chr1 20 22 1 2 0 1 0
chr1 22 30 2 1,2 1 1 0
chr1 30 32 1 2 0 1 0
chr1 32 34 1 3 0 0 1
chr1 34 247249719 0 none 0 0 0
And a header with labels. Note that if we use labels, the fourth column reports a list of labels rather than a list of file indices:
$ multiIntersectBed -i a.bed b.bed c.bed -header -names A B C
chrom start end num list A B C
chr1 6 8 1 A 1 0 0
chr1 8 12 2 A,C 1 0 1
chr1 12 15 3 A,B,C 1 1 1
chr1 15 20 2 A,B 1 1 0
chr1 20 22 1 B 0 1 0
chr1 22 30 2 A,B 1 1 0
chr1 30 32 1 B 0 1 0
chr1 32 34 1 C 0 0 1
If you are interested in an easier to follow (yet less efficient) version of the algorithm, I have posted the python prototype I developed with pybedtools.



+1. Great example of how questions on biostar are helping stimulate advances in bioinformatics technology.
agreed. it was quite fun to write.
Hi Aaron, Is the
-fparameter still functional in MultiIntersectBed, if I need a minimum overlap of 50% in all the three files.Thanks
oh cannot thanks more..
Might be a very naive question, Well I have 5 peak files and the sum of peaks is 100000 but when I use multiIntersectBed I get a total number of peaks to be 150000 why such a big difference? Does the script breaks the total regions into some bins and then makes an intersection?, if yes. what is the size of these bins?
Almost exactly what I'm looking for. Is there a way to also print out all the features from each of the 3 bed files?
Thanks.
-emptyIs multiinter strand-aware? The output doesn't give any clues whether it is!